
Abstract
Hemoglobinopathies are one of the major problems in Pakistan. A retrospective analysis of blood samples of 2731 patients from 2010 to 2014 was done at National Institute of Blood Disease & Bone Marrow Transplantation for the workup of anemia or other blood-related disorders. Whole blood samples in EDTA were collected; complete blood counts with peripheral smears were prepared. Hemoglobin (Hb) electrophoresis on Genio was performed at alkaline pH. Samples showing borderline results were further tested by high-performance liquid chromatography or for specific mutation analysis by ARMS-PCR. Out of total 2731, 935 (34.2%) patients had hemoglobinopathies. Out of these 935 patients who had hemoglobinopathies, beta thalassemia minor 51.8%, beta thalassemia major 24.1%, HbD trait 6.7, sickle/beta thalassemia 4.5%, sickle cell disease 3.9%, HbE trait 1.9%, and sickle cell trait 1.7% were most common hemoglobinopathies. Less prevalent were delta/beta thalassemia, HbE homozygous, HbD homozygous, and HbH disease.
Public Interest Statement
Hemoglobin (Hb) is a protein in red blood cells, which contains iron. It is used to transport oxygen around the human body. It carries some of the body’s respiratory carbon dioxide as well. Red blood cells get their red color from Hb. Hemoglobinopathy is a kind of genetic defect that results in abnormal structure of one of the globin chains of the Hb molecule. Hemoglobinopathies are inherited single-gene disorders and in most cases they are inherited as a dominant trait. This is a simple study in which we calculated frequency and types of hemoglobinopathies in patients of decreased Hb referred to our laboratory. Our results may play a role in arrangements at a hospital blood bank as well as in formulation of transfusion policies.
Competing Interests
The authors declare no competing interests.
1. Introduction
Hemoglobinopathies are inherited disorders of globin: the protein component of hemoglobin (Hb). Mutations in genes coding for the globin proteins that alter protein output produce the thalassemia syndromes. (Trent, Citation2006 Feb)
Hb comprises four subunits, each having one polypeptide chain and one heme group. All Hbs carry the same prosthetic heme group iron protoporphyrin IX associated with a polypeptide chain of 141 (alpha) and 146 (beta) amino acid residues. The ferrous ion of the heme is linked to the N of a histidine. The porphyrin ring is wedged into its pocket by a phenylalanine of its polypeptide chain. The polypeptide chains of adult Hb themselves are of two kinds, known as alpha and beta chains, similar in length but differing in amino acid sequence. The alpha chain of all human Hb, embryonic and adult, is the same. The non-alpha chains include the beta chain of normal adult Hb (α2β2), the gamma chain of fetal Hb (α2β2), and the delta chain of HbA2. In some variants, the gamma genes are duplicated, giving rise to two kinds of gamma chains (Marengo-Rowe, Citation2006 ).
Mutations in the globin genes can cause either a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced. Quantitative defects cause thalassemia, whereas qualitative changes, referred to as Hb variants, result in a wide range of problems including sickle cell disease, unstable Hb, decreased oxygen affinity, increased oxygen affinity and methemoglobinemia; however, the majority of qualitative mutations cause no significant change in Hb properties or clinical problems (Hoff brand, Citation2011).
According to World Health Organization (Modell, Citation2008), Hb disorders were originally endemic in 60% of 229 countries, potentially affecting 75% of births, but are now sufficiently common in 71% of countries among 89% of births either in the whole population or among minorities.
These hereditary disorders of Hb pose a massive health problem in many Third world countries including India, Pakistan, and Iran. The distribution of specific disorders varies geographically and by community (Balgir, Citation1996). WHO figures estimate that 5% of the world population is carrier for Hb disorders (World health organization–Executive board session 118th agenda point 5.2, Citation2006) causing moderate to severe hemolytic anemia leading to high degree of morbidity and mortality.
The main objective of the study was to know the frequency distribution of hemoglobinopathies in the Karachi, located in the southern part of Pakistan, the largest cosmopolitan city of the country with a population in excess of 20 million which will help in formulating various strategies for the effective control and prevention of these disorders.
2. Material and methods
This cross-sectional retrospective study included 2731 patients with the diagnosis of anemia on complete blood count (CBC) referred for screening of Hb disorders from September 2010 to December 2014 at National Institute of Blood Disease (NIBD) & Bone Marrow Transplantation—a reference laboratory which receives samples for testing and diagnosis of hemoglobinopathies from OPD and many small laboratories and clinics from all over Karachi. Institutional review board approved the study. The blood samples were collected from the patients of low Hb, with suspicion of hemoglobinopathies who visited NIBD or alternatively the samples were collected and sent by other pathology laboratories/clinics for testing of parameters like, CBC, peripheral smear, Hb analysis by electrophoresis (at alkaline pH), high-performance liquid chromatography (HPLC), and sickling test. CBCs were done by Sysmex XN 1000 analyzer, peripheral blood smears were stained with Leishman’s stain, grading of hypochromia, anisocytosis, microcytosis, macrocytosis, and polychromasia was done according to the standard criteria. Hb electrophoresis at alkaline pH was performed on Genio analyzer. HbA2 value > 3.5% was considered as cut-off point for beta thalassemia minor.
3. Results
Out of 2731 cases, 935 (34.23%) patients had hemoglobinopathies; rest of the 1792 patients had a normal Hb electrophoresis pattern. The age- and gender-wise distribution of the patients with hemoglobinopathies is described in . Majority of the patients studied were native residents of Karachi. Out of total 935 patients with hemoglobinopathies, 51.8% had β-thalassemia minor and 24.1% had β-thalassemia major, while frequency of HbD trait was 6.7%, sickle/beta thalassemia 4.5%, sickle cell disease 3.9%, HbE trait 1.9%, and sickle cell trait 1.7%, whereas less frequent hemoglobinopathies include, HbE/beta thalassemia 1.4% and HbD/beta thalassemia 1.4%, HbH disease 1.01%, HbD homozygous 0.9%. The overall frequency of various hemoglobinopathies is shown in . There was one patient with homozygous HbE disease and a single case of hereditary persistence of fetal Hb (HPFH). There were four patients with borderline results where further investigation could not be performed due to non-availability of samples.
Table 1. Age- and gender-wise distribution of patients with hemoglobinopathies
Figure 1. Frequency of various hemoglobinopathies (n = 2731).
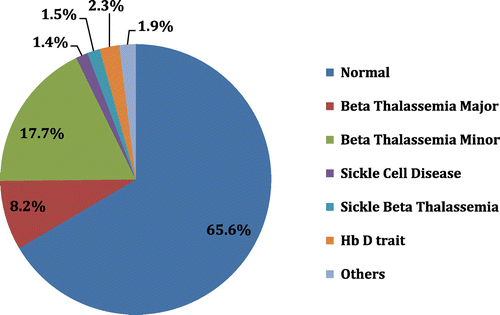
Average values of hemogram and results of Hb electrophoresis are described in and respectively.
Table 2. Hematological parameters (mean & standard deviation) in common hemoglobinopathies
Table 3. Hb electrophoresis results(mean & standard deviation) in various hemoglobinopathies
The average HbF level in beta β-thalassemia major patients was 82.67%. There was one case of HPFH with 100% HbF levels. Average HbA2 level in β-thalassemia minor patients was 4.6%, mean HbS level in sickle cell disease was 73.7%, while average HbD level in homozygous HbD (Punjab) was 96%, whereas the HbE level in HbE trait and HbE/beta thalassemia was 26.4% and 53.96%, respectively, as described in . On electrophoresis, HbE and A2 were migrated at the same position; similarly, HPLC retention time was similar for HbE and A2; therefore, HbE/beta thalassemia was diagnosed on the basis of clinical history and morphology and in some cases by DNA mutation analysis.
Majority of the patients with sickle cell disease had blood indices and blood film suggestive of hypochromic microcytic anemia. The average HbF in sickle cell patients was 28.1%. Out of 489 patients of β-thalassemia minor, 344 had MCV below 76 fl ranging from 46 to 75 fl.
4. Discussion
The overall frequency of hemoglobinopathies in this study is 34.2%, which is closer to a previous study done on 504 patients at Islamabad by U. Waheed et al. revealing a frequency of 28.4% (Waheed, Citation2012). This study also supports the finding that thalassemia is the most frequent form of hemoglobinopathy in Pakistan (Waheed, Citation2012). In another study conducted by Muhammad Saleem et al. on distribution pattern of hemoglobinopathies in northern areas of Pakistan, of the 1187 cases of refractory anemia, 305 (25.69%) has thalassemia or abnormal Hb (Saleem, Citation1985). An estimated 5000–9000 children with β-thalassemia are born per year, although no documentary registry is available in Pakistan. The estimated carrier rate is 5–7%, with 9.8 million carriers in the total population (Ahmed, Saleem, Modell, & Petrou, Citation2010).
Thalassemia is among the most common genetic disorders worldwide; 4.83% of the world’s populations carry globin chain variants, including 1.67% of the population who are heterozygous for α-thalassemia and β-thalassemia. In addition, 1.92% carry sickle Hb, 0.95% carry HbE, and 0.29% carry HbC. Thus, the worldwide birth rate of homozygous or compound heterozygous for symptomatic globin chain disorders, including α-thalassemia and β-thalassemia, is no less than 2.4 per 1000 births, of which 1.96 have sickle cell disease and 0.44 have thalassemia (Angastiniotis & Modell, Citation1998).
In Pakistan, awareness of thalassemia prevention is increasing and a new bill on thalassemia prevention and control was passed from Sindh assembly two years ago but still needs to be implemented. Our study is an attempt to determine the frequency of various hemoglobinopathies in Karachi region that can be useful in prevention and management of various hemoglobinopathies, which may play a vital role in the hospital blood bank as well as in the formulation of transfusion policies. Adequate measures and screening procedures especially prenatal diagnosis should be performed concurrently with the aim to reduce the possibility of Hb disorders in offspring, mental and physical trauma of affected patients, and socioeconomic burden of the family. Screening is affordable and an accessible way to detect carriers, and can be offered in a range of settings in different societies: In high school, before marriage, or in antenatal clinics.
Additional information
Funding
Notes on contributors
Shaista Shabbir
I, Dr Shaista Shabbir corresponding author of this article is a student of MPhil leading to PhD (Hematology) in National Institute of Blood Diseases and Bone Marrow Transplant Centre Karachi which is the first ever Pediatrics Bone Marrow Transplant Unit and Genomic Department of the country. More than 500 successful bone marrow transplants in children have been conducted by NIBD, while 3000 children in a year being suffered with bone marrow diseases. NIBD and BMT is a major referral centre for thalassemia patients; our drainage area is throughout Karachi and surroundings, so we are in a capacity to study the pattern of hemoglobinopathies in this area. Our sample will represent the true picture of whole of the disease.
References
- Ahmed, S., Saleem, M., Modell, B., & Petrou, M. (2010). Screening extended families for genetic hemoglobin disorders in Pakistan. New England Journal of Medicine, 347, 1162–1168.
- Angastiniotis, M., & Modell, B. (1998). Global epidemiology of hemoglobin disorders. Annals of the New York Academy of Sciences, 850, 251–269.10.1111/j.1749-6632.1998.tb10482.x
- Balgir, R. S. (1996). Genetic epidemiology of the three predominant abnormal hemoglobin in India. The Journal of the Association of Physicians of India, 44, 25–28.
- Hoff brand, A. (2011). Postgraduate hematology. Chi Chester, West Sussex: Wiley-Blackwell.
- Marengo-Rowe, A. J. (2006, July). Structure-function relations of human hemoglobins. In Baylor University Medical Center Proceedings, 19, 239–245.
- Modell, B. (2008). Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization, 86, 480–487.
- Saleem, M. (1985, April). Distribution pattern of haemoglobinopathies in northern areas of Pakistan. Journal of Pakistan Medical Association, 35, 106–109.
- Trent, R. J. (2006, February). Diagnosis of the haemoglobinopathies. The Clinical Biochemist Review, 27, 27–38.
- Waheed, U. (2012, December). Frequency of haemoglobinopathies: A single-centre, cross-sectional study from Islamabad, Pakistan. Eastern Mediterranean Health Journal, 18, 1257–1259.
- World health organization–Executive board session 118th agenda point 5.2. (2006). 118th session report by the secretariat on thalassemia and other haemoglobinopathies: Prevalence of haemoglobinopathies. Geneva. Retrieved 2006, May 11. 1–8.