
Abstract
Abundant knowledge is present over nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus but no attention has been given to the same aspect with type 1 diabetes mellitus (T1DM) in humans. However, various animal modeled studies are documented having induced T1DM using chemical induction by alloxan (ALX) or streptozotocin (SZ) to investigate and comprehend the nature and pathophysiology of NAFLD in T1DM. The objectives of this review were to assess whether T1DM induced by ALX or SZ could cause histological (morphological and ultrastructural) changes in rat liver to illustrate the effects of these drugs and diabetes, and pathophysiology of NAFLD in T1DM. To achieve these objectives, we systematically searched the studies from EconLit, Embase, Google Scholar, Medline, PubMed, ProQuest, Scopus, Springer Link, and Science Direct databases. Initially, 150 studies were retrieved. Out of these, 107 studies were removed as these failed to provide relevant details. Finally, 43 articles were selected for this review. This review concluded that diabetes induced by ALX or SZ, cause biochemical alterations in blood and pathophysiological variations in the liver of rats. These changes can vary from steatosis to steatohepatitis and liver fibrosis, and are similar to the modifications observed in human liver.
Public Interest Statement
Diabetes mellitus is not a single disease instead it is a syndrome having a peculiar feature of raised blood glucose level primarily due to unbalanced insulin production, secretion or action. A wealth of knowledge is present over nonalcoholic fatty liver disease and type 2 diabetes mellitus but no attention has been given to the same aspect with type 1 diabetes mellitus in humans. This review revealed that diabetes induced by ALX or SZ, cause biochemical alterations in blood and pathophysiological variations in the liver of rats. These changes can vary from steatosis to steatohepatitis and liver fibrosis, and are similar to the modifications observed in human liver.
Competing Interests
The authors declare no competing interest.
1. Introduction
Diabetes mellitus (DM) is not a single disease instead it is a syndrome having a peculiar feature of raised blood glucose level primarily due to unbalanced insulin production, secretion or action (Saltiel & Kahn, Citation2001). Recently, World Health Organization has given a report on the global burden of the disease. According to its findings the predominance of the disease in adults is assessed to be about 387 million (Alberti & Zimmet, Citation2013). Glucose is the main fuel for the body and its ultimate concentration in the blood depends upon its production and consumption by different tissues, which is rather high in case of diabetes.
Insulin is the main hormone to regulate the production and utilization of glucose by the body tissues and organs. In the absence of insulin, the liver increases glucose uptake, but kidney being insulin-independent organ, tries to cope with the higher level of glucose in case of diabetes (Baquer, Gupta, & Raju, Citation1998). Several biochemical and morphological defects occur in case of hyperglycemia due to increased glycosylation resulting in an altered protein structure. One of the major impediments of diabetes mellitus is the microvascular complications such as thickening of capillary basement membranes, retinopathy and nephropathy (Thakran, Siddiqui, & Baquer, Citation2004). If the treatment for diabetes is not started in time, the sustained hyperglycemic condition would cause serious problems such as permanent hepatic and renal deterioration. A number of experiments have been done to study the effects of hyperglycemia on the structure and function of kidney and liver (Dabroś, Kajda, & Kordowiak, Citation2001; Mifsud et al., Citation2001; Obineche et al., Citation2001).
The correlation is well established between type 2 diabetes mellitus (T2DM) and nonalcoholic fatty liver disease (NAFLD) including a large continuum of liver disorders from steatosis to cirrhosis (Ahmadieh & Azar, Citation2014; Farrell & Larter, Citation2006; Smith & Adams, Citation2011). A wealth of knowledge is presented over NAFLD and T2DM but no attention is given to the same aspect with type 1 diabetes mellitus (T1DM) in humans. However, animal modeled studies are documented having induced T1DM using chemical induction by alloxan (ALX) or streptozotocin (SZ) to investigate and comprehend the nature and pathophysiology of NAFLD in T1DM yet there are only a few studies showing the long-term effect of T1DM on the progression of liver disease (Remedio, Castellar, Barbosa, Gomes, & Caetano, Citation2011; Welt et al., Citation2004).
As the studies in the literature are of short-term follow-up of animals with T1DM, there is an ambiguity that whether the liver injury is due to drug induced T1DM or due to the toxic effects of ALX and SZ (Adewole & Ojewole, Citation2007; Can et al., Citation2005; Da̧broś, Goc, Turyna, & Kordowiak, Citation2005; Guven et al., Citation2006). So, this review assessed whether T1DM induced by ALX or SZ could cause histological (morphological and ultrastructural) changes in rat liver to illustrate the effects of these drugs and diabetes, and pathophysiology of NAFLD in T1DM.
2. Methods
A comprehensive literature search (studies published during 2001 to December 2015) was made from Embase, EconLit, Google Scholar, Medline, PubMed, ProQuest, Scopus, Springer Link and Science Direct databases. “Histological”, “Diabetes mellitus”, “Liver”, “Histopathology”, “Chronic liver disease”, “ Hepatic insufficiency”, and “Rats” were used as keywords in diverse combinations with BOOLEAN and MeSH search.
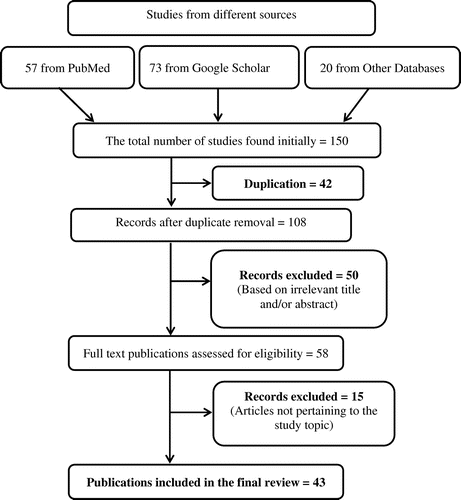
Further publications were recognized by a manual search of references of related papers, and review articles were also cited where applicable. Initially, 150 studies were retrieved. Out of these, 42 were duplicates and therefore were excluded. Out of 108 remaining articles, 50 were dropped off based on irrelevant title and/or abstract. Subsequently, full text of 58 articles were read and 15 studies were further removed as these failed to provide relevant details. Finally, 43 articles were selected for this review ().
Figure 1. Schematic diagram explaining the assortment of studies/reports.
3. Results and discussion
Clinical and experimental proofs illustrated the fact that DM has an effect on the liver along with blood vessels, kidneys, retina and nerves (Evelson et al., Citation2005; Leite et al., Citation2011; Regnell & Lernmark, Citation2011; Verderese & Younossi, Citation2013). There is a wide spectrum of clinical, metabolic and hormonal disorders that can cause NAFLD, some of which, namely obesity, malnutrition, intestinal malabsorption, dyslipidemia, thyroid disorders, and metabolic syndrome (Portincasa, Grattagliano, Palmieri, & Palasciano, Citation2005). This was the reason that DM was not predicted earlier as one of the foremost causes of chronic liver disorder.
This review illustrated that T1DM rats when given intravenous ALX showed the signs which resembled to chronic liver disease in humans, such as biochemical alterations in blood and morphological and ultrastructural lacerations in the liver. These alterations in liver started from fatty degeneration of liver cells and extended to steatohepatitis and periportal fibrosis. Characteristically, the lacerations in the liver of the diabetic animals were found to extend to all structures of the organ, involving hepatocytes, portal areas and sinusoids, intracytoplasmic organelles and nuclei, including a progressive enlargement of sinusoids, periportal fibrosis, and micro- and macrovesicular fatty degeneration and steatohepatitis.
The ultrastructural variations in hepatocytes were found in numerous experiments specifically with respect to the mitochondria, the rough endoplasmic reticulum (rER) and cell nuclei, but it is unclear that these variations were either due to T1DM state or due to the toxic effects of the drug. Although ALX and SZ are used to induce T1DM by destructing pancreatic beta cells, but they also elicit toxic effects on the kidneys and livers of several species (Junod et al., Citation1967).
There are a number of factors which play a role in the systemic toxicity of these drugs and include species, age, body weight of the animals under experiment, the hydration status, route through which the drug is administered, the rate at which drug is being infused and duration of fasting for drug administration (Lucchesi, Cassettari, & Spadella, Citation2015). Systemic noxious effects of ALX and SZ are usually seen within the first 2 weeks after diabetes induction. Peculiar concern is needed with ALX as it is a drug with narrow therapeutic index and expiries are usually seen as a result of minor fluctuations in doses mainly due to diabetic ketoacidosis and/or kidney or liver failure (Lucchesi et al., Citation2015). With the knowledge of aforementioned points it is necessary to observe the animal in the acute or subacute phases of ALX action as the actual cause of the changes in the liver can then be identified.
A study performed by Kume, Ohmachi, Itagaki, Tamura, and Doi (Citation1994) on diabetic mice showed acute morphological alterations in the liver involving hepatocyte hypertrophy, bile duct hyperplasia and an increased number of intracytoplasmic acidophilus pellets. All of these findings suggest that the alteration in the structure and functions of liver in diabetic mice are due to the toxic effects of SZ rather to the effect of DM itself.
A study performed by Voss, Brachmann, and Hartmann (Citation1988) revealed a detrimental effect of SZ on the rat liver as evident by the elevated levels of ALT in the blood, even when DM was not present yet. On the other hand, the researchers observed that this phenomenon was of lesser duration, i.e. of 10 days and was longer i.e. of 30 days only in animals who had hyperglycemic condition upon the induction of DM. Serum levels of both AST and ALT remained elevated for the first 2 weeks of treatment and just ALT level persisted considerably raised at 6, 14 and 26 weeks after the induction of diabetes. However, some other authors stated their results differently (Kume et al., Citation1994; Mahmoud & Al-Salahy, Citation2004), stating they did not notice any substantial changes in other liver function tests, containing liver enzymes of excretion, cholesterol, triglycerides, and low and high density lipoproteins at any time point of their study. Two scientists namely Kozyritskij and Minchenko witnessed substantial ultrastructural modifications in hepatocytes of alloxan-induced diabetic rats after a 12-week period, involving decrease in the development of the rER, proliferation in mitochondrial size and no increase of smooth endoplasmic reticulum elements (Kozyritskiĭ & Minchenko, Citation1977). Astonishingly, upon the administration of insulin and treatment of hyperglycemic condition, all of the ultra-structural abnormalities in the rats’ liver were restored. Now this discovery supports that all of the abnormalities observed in the structure and function of liver were due to hyperglycemia of DM, especially after the 2nd week of diabetes, and not just the toxicity of ALX and SZ. Successive studies proved this postulate by restoring the liver functions back to normal through effective control over hyperglycemic condition (Da̧broś et al., Citation2005; Lucchesi et al., Citation2015; Mahmoud & Al-Salahy, Citation2004).
An issue of interest is that some of the liver lacerations which are caused by ALX and SZ in the acute and sub-acute phases of treatment are also seen in chronic stages of diabetes induction. This proposes that these liver lacerations may result from separate effects of these chemicals or chronic hyperglycemia and the combined action of these factors. This postulate was proposed by Laguens, Candela, Hernández, and Gagliardino (Citation1980) as they observed a distention of cisterns and degranulation of the rER surface, mitochondrial inflammation and a loss of mitochondrial cristae were witnessed in hepatocytes of C3H-s mice, 21 days after SZ treatment both in animals that developed diabetes and in those that did not develop the disease. To test the hypothesis that whether hyperglycemia plays a significant role in the genesis of liver pathology, two researchers Mahmoud and Al-Salahy (Citation2004) designed a test in which they used fish as experimental animal. Repeated doses of glucose were administered to one group and ALX was given to other group and liver injury was studied indicating an enlargement of sinusoids, dilation of cisterns and a loss of rER ribosomes in the hepatocytes. Then they treated the hyperglycemia using an aqueous suspension of lupine seeds and demonstrated that the liver histology and functions of liver were back to normal again. There is a close relationship in the sternness of the liver lacerations with the time course of diabetes; severity worsens with the duration of the experimental follow-up, especially in diabetic animals after 26 weeks of an uncontrolled hyperglycemic state. Even in the 6th week of diabetes, enlargement of sinusoids and focal microvesicular fatty degeneration with vacuolization of liver cells was seen. With the advent of 14th week, those initial lacerations progressively extend to the rest of the liver parenchyma and by 26th week of diabetes, the whole organ was invaded, when the liver characteristically showed diffuse macrovesicular fatty degeneration that was sometimes supplemented by interstitial mononuclear inflammatory infiltrates (steatohepatitis) and mild periportal fibrosis. The last two variations were only observed in animals immolate at 26 weeks of diabetes, this pinpoints that the progression of liver fatty degeneration to steatohepatitis and fibrosis is reliant on the prolong contact of liver cells to hyperglycemic condition.
The liver of diabetic animals was also presented with ultrastructural progressive injury which are considered mainly to be due to the incompetence of the liver cell ultrastructure, micro and macrovesicular fatty degeneration, loss of intracytoplasmic organelles, and qualitative and particular variations of intracytoplasmic organelles and nuclei, which is noticeable as a lack of rER development, an obvious reduction of mitochondrial crests, mitochondrial inflammation and an abnormal distribution of heterochromatin granules. Welt et al. (Citation2004) stated related outcomes using morphological and morphometric examination of liver tissue from Wistar rats which were exposed to SZ to induce diabetes or exposed to acute hypoxia over a period of 16 weeks. They detected a surge in the size of hepatocytes and their nuclei, a decrease number of rER cisterns, a reduced nucleus/cytoplasm ratio and hepatic glycogen content, and the existence of various fat vacuoles in liver cells (“Ito cells”) with a high density of types I, III and VI collagens around the portal vessels and within the space of Disse, which indicated hepatic fibrosis. DM also has a negative effect on liver cell nuclei as stated by Doi, Yamanouchi, Kume, and Yasoshima (Citation1997) exhibiting hepatocyte nuclei duplicate and heterogeneity in shape, size, and distribution of nuclear chromatin granules along with the enlarged area of hepatocytes.
Guven et al. (Citation2006) performed an experiment over a period of 4 weeks to inspect whether melatonin protects lacerated liver as caused by toxic effects of SZ to induce DM and stated the following outcomes. There was hydropic degeneration, sinusoid congestion, focal necrosis and microvesicular fatty infiltration of hepatocytes that was accompanied with moderate periportal swelling, a decline number of intracytoplasmic organelles, an increase of a homogeneous substance in nuclear chromatin and mitochondrial deterioration. Nevertheless, these researchers witnessed no macrovesicular fatty deterioration or hepatic fibrosis which indicates the chronic nature of these illnesses. Adewole and Ojewole (Citation2007) were reviewing the hypoglycemic effects of herbal aqueous extract of Artocarpus communis. They observed and stated the same pathophysiological changes in the liver of SZ induced DM animals as narrated before.
Remedio et al. (Citation2011) performed a study to check the effects of exercise on the rat liver morphology in T1DM induced by ALX. The follow-up period was of 8 weeks and he observed very favorable and beneficial results as there was significant increase in the number of mitochondria in the liver cells of sedentary diabetic animals in contrast to inactive controls with no proof of any remarkable ultrastructural variations in these organelles.
It is generally accepted that long-term DM does affect liver as it affect other organs. The mechanism by which T2DM damage liver is well understood but in case of T1DM, it is not documented. Cellular oxidative stress (OS) is thought to play a major part in the genesis and advancement of chronic diabetic lacerations on vessels, kidneys, retina, nerves, and possibly the liver of diabetic humans and animals (Evelson et al., Citation2005; Kaneto et al., Citation2006; Khan, Farhangkhoee, & Chakrabarti, Citation2006; Roskams et al., Citation2003). One of the prime factors for the progression of cirrhosis in diabetic patients with NAFLD is the persistence of a diabetic ailment and chronic stress in liver cells specifically in obese patients as proved by Mendez-Sanchez, Chavez-Tapia, and Uribe (Citation2003). When liver oxidizes a large amount of free fatty acids (FFAs), it also produces reactive oxygen species (ROS) which cause lipid peroxidation and physical and metabolic variations in the cells and cell death. Although body has a defensive mechanism in the form of anti-oxidants which defend the body from the detrimental effects of such ROS yet this balance is broken down in case of DM which actually starts oxidative process. In case of DM related oxidative stress sustained elevated levels of blood glucose initiate oxidative stress which causes the liver to adapt to cope with this stressful condition resulting in the activation and/or inhibition of various molecular sites that transduce and transcribe signals which control the biological cell cycle (Yang, Lin, Mandal, Huang, & Diehl, Citation2001). These changes enable the liver to reproduce and regenerate, which result in apoptosis or cell death. It is also observed that when only FFA is present in large amount, apoptosis of liver cells is initiated thus pioneering one of the mechanisms of liver injury as commonly seen in patients of NAFLD (Wigg et al., Citation2001).
The long term effects of ALX in rats’ liver were observed in a study (Lucchesi, Freitas, Cassettari, Marques, & Spadella, Citation2013) and it was concluded that T1DM altered the oxidative balance in the liver which was evident by a substantial rise in ROS in liver tissue and a significant decline in the defense antioxidants. These facts proved that the pathophysiological changes observed in the liver and the alterations in the liver function markers i.e. AST and ALT, are all due to the effect of DM-related chronic stress in liver.
Regnell and Lernmark (Citation2011) then step forward and proposed three main mechanisms which either individually or in association with each other are involved in the pathogenesis of NAFLD in patients with T1DM. The three mechanisms are (1) atypical rates or malfunction of lipoproteins would create variations in triglyceride secretion from liver cells and cause an accumulation of fat in the liver; (2) triggering of transcription factors of hepatic metabolism by hyperglycemia, such as sterol regulatory element-binding proteins (SREBPs) and the carbohydrate response element-binding protein (ChREBP), would stimulate hepatic lipogenesis; and (3) surplus liver glucose transported by the glucose transporter 2 factor (GLUT2) would result in intrahepatic fat synthesis.
Various modifications are observed in the liver of T1DM patients both of qualitative and quantitative nature, though they were having astringent metabolic control, namely high proportion of triglycerides, and low and very low density lipoproteins (LDL; VLDL), cholesterol, alterations in the structure of the peripheral layers of lipoproteins, LDL oxidation, apolipoprotein glycation, and the amount of small dense particles (Vergès, Citation2009). The changes in lipoproteins would lead to change in the utility of these substances ultimately resulting in NAFLD.
Anyways, hyperglycemia alone does not fully elaborate all the variations. Even peripheral hyperinsulinemia which is usually caused by continuous administration of subcutaneous insulin in patients with T1DM. It also play its part in the pathophysiological modifications and ultimate accumulation of fats in the liver (Vergès, Citation2009). Likewise, it may also be possible that NAFLD is not the resultant disorder rather it is causative to the lipoprotein abnormalities witnessed in those patients (Lenaerts, Verresen, Steenbergen, & Fevery, Citation1990). SREBPs are mainly involved in the process of transcription which regulate the expression of over 30 genes related to the synthesis and release of cholesterol, fatty acids, triglycerides, and phospholipids (Horton, Goldstein, & Brown, Citation2002). In vitro experiments showed that raised concentrations of blood glucose raise the concentrations of SREBP-1c, a precursor of SREBPs, in the endoplasmic reticulum. SREBP-1c acts with ChREBP to surge the expression of lipogenic genes derived from pyruvate kinase (LPK) and the enzymes acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) (Ferré & Foufelle, Citation2007). LPK catalyzes the transformation of phosphoenolpyruvate to pyruvate, which is the prime source of ACC that is chiefly used in the production of fatty acids (Browning & Horton, Citation2004). FAS catalyzes the transformation of malonyl-CoA into long chain fatty acids (Weickert & Pfeiffer, Citation2006). ChREBP can stimulate the transcription of genes which activate LPK as a result of hyperglycemia, even without the contribution of insulin as proposed by Yamashita et al. (Citation2001). So ChREBP is the prime causative factor to the building up of fats in the liver of T1DM patients (Elasha, Alzahrani, Almalki, Alomair, & Alshakweer, Citation2010).
The third mechanism responsible for NAFLD is facilitated by GLUT2 and controlled by SREBP1-c. GLUT2 is principally present in liver, gut, pancreatic β-cells and kidney and is a facilitative glucose transporter on target cells (Thorens, Sarkar, Kaback, & Lodish, Citation1988). In rats hyperglycemia and hypoinsulinemia are the prime reasons for activating hepatic expression of GLUT2 (Postic et al., Citation1993). In case of DM, an excess amount of glucose reaches to liver via blood which results in an excess accumulation of glucose in liver cells which in turn promptly transform this excess glucose into fatty acid deposits. Now, these fatty acids are either transfused into the blood in the form of lipoproteins or they are stockpiled in the liver parenchyma in the form of triglycerides, which cause in steatosis.
Insulin also plays its part in causing steatosis as demonstrated by Regnell and Lernmark (Citation2011). They performed this study in rodents and observed that the flow of FFAs was modified by insulin as it is transported by protein pathways mediated by 2 and 5 FATPs (fatty acid transport proteins). The actions of 2 and 5 FATP are dose-dependent and bimodal, in case of hypoinsulinemia they are mediated by insulin receptor substrate-2 (IRS-2) and in case of hyperinsulinemia; via insulin receptor substrate-1 (IRS-1). In the light of above discussion, it is concluded that insulin is required in perfect range for the proper fat metabolism by liver. Any alterations in the insulin concentration of blood as in case of a decrease in production such as in T1DM or excess plasma level on insulin as in T2DM or improper exogenous insulin administration, all cause a buildup of fat in liver (Lucchesi et al., Citation2015).
Anyhow, the development of NAFLD to nonalcoholic steatohepatitis (NASH) followed by fibrosis and/or cirrhosis in case of T1DM is not proved yet. A number of hypothesis suggested for the dawn of these problems in T2DM patients, which are present in T1DM patients, may be proposed as a great portion of these patients are also obese, and mechanisms of peripheral insulin resistance and endogenous or iatrogenic hyperinsulinemia are also witnessed (Mendez-Sanchez et al., Citation2003).
The onset and development of NAFLD to NASH and liver cirrhosis involves two essential factors as proposed by Portincasa et al. (Citation2005). The first factor comprises patients’ risk factors, namely lifestyle and genetic defects, and the second factor include alterations in intra- and extracellular molecular mechanisms which cause liver damage, comprising of FFA oxidation, hepatocellular necrosis and fibrosis, lipid peroxidation, ROS formation and proinflammatory cytokine release, which is responsible for inflammation. The chances of liver damage increase due to the accumulation of triglycerides in it as stated by Dowman, Tomlinson, and Newsome (Citation2010), basically due to inflammatory cytokines/adipocytokines or hepatocellular oxidative stress, by favoring the progression of NAFLD to NASH and/or fibrosis. These researchers advocate that FFAs also act directly on the liver to stimulate liver injury. These compounds undergo β-oxidation, got esterified with glycerol to formtriglycerides, ultimately resulting in the accumulation of fat in the liver. It is proposed that FFAs promote the NAFLD by surging hepatic oxidative stress and/or activating numerous inflammatory pathways.
According to Roskams et al. (Citation2003), as hepatocytes have a power to regenerate, the dead liver cells in healthy persons kindle replication of mature hepatocytes to rebuild the impaired liver tissue but in case of NAFLD, the oxidative stress prevents the replication of mature hepatocytes leading to the expansion of hepatic progenitors (oval cells) which then differentiate into “hepatocyte-like cells” but are not hepatocytes. As the condition of NAFLD worsens, there is an increase in the production of oval cells and “hepatocyte-like cells” ultimately leading to hepatic fibrosis and hepatocellular carcinogenesis.
4. Conclusions
Diabetes induced by ALX or SZ cause biochemical alterations in blood and pathophysiological variations in the liver of rats. These changes can vary from steatosis to steatohepatitis and liver fibrosis and are similar to the modifications observed in human liver. The extent of morphological and ultrastructural lacerations in liver are closely related to the timing of diabetes; in case of extended follow-up period particularly in diabetic animals after 26 weeks of uncontrolled hyperglycemia, the laceration observed were more severe. This review of diabetic chronic liver disease may serve as a basis for new investigations.
Additional information
Funding
Notes on contributors
Muhammad Rehan Sarwar
We are a team of clinical medicine and pharmacy practice researchers. Affiliated with Nishtar Medical College and Department of Pharmacy, The Islamia University of Bahawalpur, Pakistan. Our research primarily focuses on health literacy, drug utilization, and chronic and infectious diseases (e.g. diabetes, cancer, epilepsy, migraine, tuberculosis, etc.).
References
- Adewole, S. O., & Ojewole, J. A. (2007). Artocarpus communis Forst. Root-bark aqueous extract- and strepto-zotocin-induced ultrastructural and metabolic changes in hepatic tissues of wistar rats. African Journal of Traditional, Complementary and Alternative Medicines, 4, 397–410.
- Ahmadieh, H., & Azar, S. T. (2014). Liver disease and diabetes: Association, pathophysiology, and management. Diabetes Research and Clinical Practice, 104, 53–62.10.1016/j.diabres.2014.01.003
- Alberti, K. G. M., & Zimmet, P. (2013). Epidemiology: Global burden of disease—where does diabetes mellitus fit in? Nature Reviews Endocrinology, 9, 258–260.10.1038/nrendo.2013.54
- Baquer, N. Z., Gupta, D., & Raju, J. (1998). Regulation of metabolic pathways in liver and kidney during experimental diabetes: Effects of antidiabetic compounds. Indian Journal of Clinical Biochemistry, 13, 63–80.10.1007/BF02867866
- Browning, J. D., & Horton, J. D. (2004). Molecular mediators of hepatic steatosis and liver injury. Journal of Clinical Investigation, 114, 147–152.10.1172/JCI200422422
- Can, B., Ulusu, N. N., Kilinç, K., Leyla Acan, N., Saran, Y., & Turan, B. (2005). Selenium treatment protects diabetes-induced biochemical and ultrastructural alterations in liver tissue. Biological Trace Element Research, 105, 135–150.10.1385/BTER:105:1-3
- Da̧broś, W., Goc, A., Turyna, B., & Kordowiak, A. (2005). Sodium metavanadate affected control and streptozotocin-diabetic rat liver golgi complexes. Polish Journal of Pathology: Official Journal of the Polish Society of Pathologists, 57, 91–97.
- Dabroś, W., Kajda, B., & Kordowiak, A. M. (2001). Control and STZ-diabetic rat liver Golgi complexes under the influence of bis (2, 2′-bipyridine) oxovanadium (IV) sulphate. The morphological investigation. Polish Journal of Pathology: Official Journal of the Polish Society of Pathologists, 53, 123–128.
- Doi, K., Yamanouchi, J., Kume, E., & Yasoshima, A. (1997). Morphologic changes in hepatocyte nuclei of streptozotocin (SZ)-induced diabetic mice. Experimental and Toxicologic Pathology, 49, 295–299.10.1016/S0940-2993(97)80041-3
- Dowman, J. K., Tomlinson, J., & Newsome, P. (2010). Pathogenesis of non-alcoholic fatty liver disease. QJM, 103, 71–83.10.1093/qjmed/hcp158
- Elasha, H., Alzahrani, S., Almalki, M., Alomair, A., & Alshakweer, W. (2010). Acute hepatic steatosis: An unusual manifestation of poorly controlled type 1 diabetes. Practical Diabetes International, 27, 282–280a.10.1002/pdi.1503
- Evelson, P., Susemihl, C., Villarreal, I., Llesuy, S., Rodríguez, R., Peredo, H., … Filinger, E. (2005). Hepatic morphological changes and oxidative stress in chronic streptozotocin-diabetic rats. Annals of Hepatology, 4, 115–120.
- Farrell, G. C., & Larter, C. Z. (2006). Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology, 43(S1), 99–112.
- Ferré, P., & Foufelle, F. (2007). SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Hormone Research in Paediatrics, 68, 72–82.10.1159/000100426
- Guven, A., Yavuz, O., Cam, M., Ercan, F., Bukan, N., Comunoglu, C., & Gokce, F. (2006). Effects of melatonin on streptozotocin-induced diabetic liver injury in rats. Acta Histochemica, 108, 85–93.10.1016/j.acthis.2006.03.005
- Horton, J. D., Goldstein, J. L., & Brown, M. S. (2002). SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. Journal of Clinical Investigation, 109, 1125–1131.10.1172/JCI0215593
- Junod, A., Lambert, A., Orci, L., Pictet, R., Gonet, A., & Renold, A. (1967). Studies of the diabetogenic action of streptozotocin. Experimental Biology and Medicine, 126, 201–205.10.3181/00379727-126-32401
- Kaneto, H., Nakatani, Y., Kawamori, D., Miyatsuka, T., Matsuoka, T.-A., Matsuhisa, M., & Yamasaki, Y. (2006). Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic β-cell dysfunction and insulin resistance. The International Journal of Biochemistry & Cell Biology, 38, 782–793.10.1016/j.biocel.2006.01.004
- Khan, Z. A., Farhangkhoee, H., & Chakrabarti, S. (2006). Towards newer molecular targets for chronic diabetic complications. Current Vascular Pharmacology, 4, 45–57.10.2174/157016106775203081
- Kozyritskiĭ, V., & Minchenko, A. (1977). Ultrastructural changes in rat hepatocytes with diabetes and insulin administration. TSitologiia i genetika, 12, 397–401.
- Kume, E., Ohmachi, Y., Itagaki, S.-I., Tamura, K., & Doi, K. (1994). Hepatic changes of mice in the subacute phase of streptozotocin (SZ)-induced diabetes. Experimental and Toxicologic Pathology, 46, 368–374.10.1016/S0940-2993(11)80119-3
- Laguens, R., Candela, S., Hernández, R., & Gagliardino, J. (1980). Streptozotocin-induced liver damage in mice. Hormone and Metabolic Research, 12, 197–201.10.1055/s-2007-996241
- Leite, N. C., Villela-Nogueira, C. A., Pannain, V. L., Bottino, A. C., Rezende, G. F., Cardoso, C. R., & Salles, G. F. (2011). Histopathological stages of nonalcoholic fatty liver disease in type 2 diabetes: Prevalences and correlated factors. Liver International, 31, 700–706.10.1111/liv.2011.31.issue-5
- Lenaerts, J., Verresen, L., Steenbergen, W., & Fevery, J. (1990). Fatty liver hepatitis and type 5 hyperlipoproteinemia in juvenile diabetes mellitus. Journal of Clinical Gastroenterology, 12, 93–97.10.1097/00004836-199002000-00024
- Lucchesi, A. N., Freitas, N. T. D., Cassettari, L. L., Marques, S. F. G., & Spadella, C. T. (2013). Diabetes mellitus triggers oxidative stress in the liver of alloxan-treated rats: A mechanism for diabetic chronic liver disease. Acta Cirurgica Brasileira, 28, 502–508.10.1590/S0102-86502013000700005
- Lucchesi, A. N., Cassettari, L. L., & Spadella, C. T. (2015). Alloxan-induced diabetes causes morphological and ultrastructural changes in rat liver that resemble the natural history of chronic fatty liver disease in humans. Journal of Diabetes Research, 2015, 1–11.
- Mahmoud, A., & Al-Salahy, M. (2004). Physiological and histological studies on the effect of lupine seeds on the fish, clarias lazera treated with glucose and alloxan. Fish Physiology and Biochemistry, 30, 213–229.10.1007/s10695-005-8243-6
- Mendez-Sanchez, N., Chavez-Tapia, N., & Uribe, M. (2003). An update on non-alcoholic fatty liver disease. Revista de investigacion clinica; organo del Hospital de Enfermedades de la Nutricion, 56, 72–82.
- Mifsud, S., Allen, T., Bertram, J., Hulthen, U., Kelly, D., Cooper, M., … Gilbert, R. (2001). Podocyte foot process broadening in experimental diabetic nephropathy: Amelioration with renin-angiotensin blockade. Diabetologia, 44, 878–882.
- Obineche, E., Mensah-Brown, E., Chandranath, S., Ahmed, I., Naseer, O., & Adem, A. (2001). Morphological changes in the rat kidney following long-term diabetes. Archives of Physiology and Biochemistry, 109, 241–245.10.1076/apab.109.3.241.11592
- Portincasa, P., Grattagliano, I., Palmieri, V. O., & Palasciano, G. (2005). Nonalcoholic steatohepatitis: Recent advances from experimental models to clinical management. Clinical Biochemistry, 38, 203–217.10.1016/j.clinbiochem.2004.10.014
- Postic, C., Burcelin, R., Rencurel, F., Pegorier, J.-P., Loizeau, M., Girard, J., & Leturque, A. (1993). Evidence for a transient inhibitory effect of insulin on GLUT2 expression in the liver: Studies in vivo and in vitro. Biochemical Journal, 293, 119–124.10.1042/bj2930119
- Regnell, S. E., & Lernmark, A. (2011). Hepatic steatosis in type 1 diabetes. The Review of Diabetic Studies, 8, 454–467.10.1900/RDS.2011.8.454
- Remedio, R. N., Castellar, A., Barbosa, R. A., Gomes, R. J., & Caetano, F. H. (2011). Morphology and protein content of hepatocytes in type I diabetic rats submitted to physical exercises. Micron, 42, 484–491.10.1016/j.micron.2011.01.008
- Roskams, T., Yang, S. Q., Koteish, A., Durnez, A., DeVos, R., Huang, X., … Diehl, A. M. (2003). Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. The American Journal of Pathology, 163, 1301–1311.10.1016/S0002-9440(10)63489-X
- Saltiel, A. R., & Kahn, C. R. (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 414, 799–806.10.1038/414799a
- Smith, B. W., & Adams, L. A. (2011). Nonalcoholic fatty liver disease and diabetes mellitus: Pathogenesis and treatment. Nature Reviews Endocrinology, 7, 456–465.10.1038/nrendo.2011.72
- Thakran, S., Siddiqui, M., & Baquer, N. Z. (2004). Trigonella foenum graecum seed powder protects against histopathological abnormalities in tissues of diabetic rats. Molecular and Cellular Biochemistry, 266, 151–159.10.1023/B:MCBI.0000049153.14295.0d
- Thorens, B., Sarkar, H. K., Kaback, H. R., & Lodish, H. F. (1988). Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and β-pancreatic islet cells. Cell, 55, 281–290.10.1016/0092-8674(88)90051-7
- Verderese, J. P., & Younossi, Z. (2013). Interaction of Type 2 diabetes and nonalcoholic fatty liver disease. Expert Review of Gastroenterology & Hepatology, 7, 405–407.10.1586/17474124.2013.811047
- Vergès, B. (2009). Lipid disorders in type 1 diabetes. Diabetes & Metabolism, 35, 353–360.10.1016/j.diabet.2009.04.004
- Voss, C., Brachmann, K., & Hartmann, K. (1988). Effect of streptozotocin on transaminases, creatinine and urea in serum of rats. Experimental and Clinical Endocrinology & Diabetes, 92, 37–42.10.1055/s-0029-1210779
- Weickert, M., & Pfeiffer, A. (2006). Signalling mechanisms linking hepatic glucose and lipid metabolism. Diabetologia, 49, 1732–1741.10.1007/s00125-006-0295-3
- Welt, K., Weiss, J., Martin, R., Dettmer, D., Hermsdorf, T., Asayama, K., … Fitzl, G. (2004). Ultrastructural, immunohistochemical and biochemical investigations of the rat liver exposed to experimental diabetes und acute hypoxia with and without application of Ginkgo extract. Experimental and Toxicologic Pathology, 55, 331–345.10.1078/0940-2993-00337
- Wigg, A., Roberts-Thomson, I., Dymock, R., McCarthy, P., Grose, R., & Cummins, A. (2001). The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut, 48, 206–211.10.1136/gut.48.2.206
- Yamashita, H., Takenoshita, M., Sakurai, M., Bruick, R. K., Henzel, W. J., Shillinglaw, W., … Uyeda, K. (2001). A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proceedings of the National Academy of Sciences, 98, 9116–9121.10.1073/pnas.161284298
- Yang, S. Q., Lin, H. Z., Mandal, A. K., Huang, J., & Diehl, A. M. (2001). Disrupted signaling and inhibited regeneration in obese mice with fatty livers: Implications for nonalcoholic fatty liver disease pathophysiology. Hepatology, 34, 694–706.10.1053/jhep.2001.28054