
Abstract
Beta-thalassemia (β-thal) is an inherited hemoglobin disorder, characterized by the absence of or reduced hemoglobin chains that result in microcytic hypochromic anemia. In this case, we describe a patient case originating from Syria, and his hematology data were (Hb A1 = 12.5%, Hb F = 83.7, Hb A2 = 3.8%). The molecular analysis based on direct sequencing of the β-globin gene showed a rare combined heterozygous IVS-I-1 (G>A) (HBB:c.92+1 G>A)/−29 A > G (HBB:c.−79 A>G) causing β°/β+ thalassemia intermedia. Polymerase chain reaction–restriction fragment length (PCR–RFLP) method revealed that the patient had a homozygous (TT) for Xmn-1 locus. Our result showed the presence of rare β-thal (−29 A>G) mutation associated with IVS-I-1 (G>A). Our finding suggests the presence of this mutation resulted from old migrations.
PUBLIC INTEREST STATEMENT
Beta-thalassemia is one of the major types of thalassemia, and it results from a decrease in lack of beta-globin chain production. It is widespread in the Mediterranean area, Middle East, South Asia, South China, India, and Africa. Beta thalassemia is classified into three types depending on the severity of symptoms: minor, intermedia, and major. Most of the beta-thalassemia mutations are caused by point mutations. In Syrian population, till date, 38 common mutations were detected. Here we presented a new case in a Syrian family that had two male patients with a compound heterozygous of two mutations IVS-I-1 (G>A) (HBB:c.92+1 G>A)/−29 A>G (HBB:c.−79 A>G) (β°/β+) on the β-globin gene causing β-thal intermedia phenotype. To the best of our knowledge, the (−29 A>G) mutation is rare in Syrian population, and this compound heterozygous genotype was not previously reported.
Competing Interests
The authors declare no competing interests.
1. Introduction
Beta-thalassemia (β-thal) is one of the major types of thalassemia, and it results from a decrease in lack of beta-globin chain production. The disease is widespread in the Mediterranean area, Middle East, South Asia, South China, India, and Africa (Weatherall & Clegg, Citation2001). β-thal is classified into three types depending on the severity of symptoms: minor, intermedia, and major (Cao & Galanello, Citation2010; Cao, Moi, & Galanello, Citation2011). At the molecular level, the disease is very heterogeneous. There are over 924 different genomic alterations related to the β-globin gene that are described in the human hemoglobin (Hb) variant (HbVar) database (Hardison et al., Citation2002). According to the literature, population studies indicate that 40 mutations account for 90% or more of the β-thal worldwide (Cao & Galanello, Citation2010; Thein, Citation2013). Most of the β-thal mutations are caused by point mutations, small deletions or insertions within the coding regions, the exon-intron junctions, and substitution within the TATA box of the β-globin gene promoter. The types of these mutations are typically ethnic-specific (Giardine et al., Citation2014). In Syrian population, till date, 38 common mutations were detected (Murad et al., Citation2018); other mutations were rarely identified.
Here we presented a rare case in a Syrian family that had two male patients with a compound heterozygous of two mutations IVS-I-1 (G>A) (HBB:c.92 + 1 G>A)/−29 A>G (HBB:c.−79 A>G) (β°/β+) on the β-globin gene causing β-thal intermedia phenotype. To the best of our knowledge, the (−29 A>G) mutation is rare in Syrian population, and this case was not previously reported.
2. Methods: Case presentation
A 16-month-old male child, the second child of a family, had β-thal history referred to the Syrian thalassemia center in Damascus in February 2009. His parents were non-consanguineous; his mother was 39 and father was 43 years old when he was born. The infant was born in the 37th week of gestation with a birth weight of 2.5 kg. A clinical history revealed an explain cry pallor, anemia, delay walking, and upper respiratory tract infection. Hematological parameters of the parents and the patient case were obtained with an automated differential cell counter (ABX Micros ES60; HORIBA ABX SAS, Montpellier, France). Capillary hemoglobin electrophoresis (Hb) analysis was measured using the Capillarys 2 system (Sebia, Lisses, France). Hb level was Hb A1 = 12.5%, Hb F = 83.7%, Hb A2 = 3.8%. Peripheral blood test showed red blood cell (RBC) count was (3.36 × 1012/L), Hb level was 8.1 g/dl, the mean corpuscular volume (MCV) was 70.5 fL, mean corpuscular Hb (MCH) was 22.3 pg, mean corpuscular Hb concentration (MCHC) was 31.6%, and red cell distribution width (RDW) was 18.2%. After that, the patient was started with erythrocyte transfusion periodicity. Recently (at age 8 years), the patient case has splenomegaly (15 cm). The first old son had the same clinical features, and he died at age 25 years. Written informed consent for publication of their clinical details was obtained from the parent.
Genomic DNA was isolated from peripheral blood from the parents and child using the QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacture's instructions. Purified gDNA was run on a 0.8% agarose gel. The quality and quantity of the DNA was determined spectrophotometrically (NanoVue™; GE HealthCare, Freiburg, Germany). The suitable primers were used for three exons of the β-globin gene including the promoter, first intron, 5′ and 3′ untranslated region (UTR) sequences as previously reported (Yassin, Sirdah, Al Haddad, Lubbad, & Al-Yazji, Citation2013). Detection of Xmn-I locus was performed with polymerase chain reaction–restriction fragment length (PCR-RFLP) technique with specific primers and restriction enzyme Xmn-I (Rujito et al., Citation2016). Reverse hybridization assay (α-Globin StripAssay® 4–160; ViennaLab Diagnostics Gmb Vienna, Austria) which covers 21 of α-thal mutations was used according to the manufacturer’s instructions.
Direct DNA sequencing of the entire human HBB gene was done on an ABI PRISM 310-DNA Analyzer (Applied Biosystem, Foster City, CA, USA) as previously reported (Murad, Moassas, Jarjour, Mukhalalaty, & Al-Achkar, Citation2014).
3. Results
In this case, the blood and physical examination of the patient showed that he was affected by thalassemia intermedia and he had anemia and pallor; the hematological and molecular data of the family are described in Table . Moreover, the results of Capillary electrophoresis (CE) showed Hb A = 95.1%, Hb A2 = 4.9% for the mother and Hb A = 12.5%, Hb A2 = 3.8%, Hb F = 83.7 for the child (Figure ). Direct DNA sequencing for the tree exons of β-globin gene revealed that the mother carried (−29 A>G) mutation in a heterozygous state and the father carried the (IVS-I-1 G>A) mutation in a heterozygous state, while the child carried acompound heterozygous genotype of two mutations IVS-I-1 (G>A) (HBB:c.92+1 G>A)/-29 A>G (HBB:c.-79 A>G) (β°/β+). On the other hand, the results of the α-thal test for the parent and the patient case revealed that none of the 21 common mutations covered by the kit were present. The result of PCR/RFLP indicates that, in our patient case, the homozygosity (TT) genotype of Xmn-I polymorphism at −158 to the Gγ-globin gene was observed.
Table 1. The hematological and molecular data of the family.
Figure 1. Direct sequencing analysis revealed the PCR fragment on the β-globin gene. (A) and (C): The arrows indicate the G>A substitution at position −29 in the child and the mother, respectively; (B) and (D): The arrows indicate the G>A substitution at position IVS-I-1 in the child and the father, respectively.
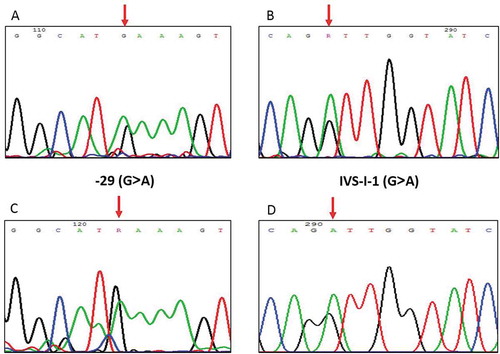
4. Discussion
Syria has a special geographical location, which has been in contact with various races and ethnic groups (Beshara, Citation2011). Syrian population is very heterogeneous, so a total of 38 different mutations of β-thal have been previously identified (Murad et al., Citation2018).
Here we report a rare compound heterozygous IVS-I-1 (G>A)/-29 A>G causing β°/β+ thalassemia intermedia in a Syrian family.
The β°-thal HBB:c.92+1 G>A mutation acts by altering the conserved dinucleotide GT present in the consensus donor sequence of the first intron of the β-globin gene (Tadmouri & Gulen, Citation2003). This mutation is one of the common mutation among β-globin gene, and it was described in many populations with different frequency (Indrak et al., Citation1992; Tadmouri & Gulen, Citation2003; Villegas, Ropero, Gonzalez, Anguita, & Espinos, Citation2001). In our previous study, this mutation was the second common mutation in Syria (Jarjour, Murad, Moasses, & Al-Achkar, Citation2014).
However, the HBB:c.−79 A>G substitution (A>G at position −29) is within the TATA “box”. This position is thought to be an essential part of the promoter unit in eukaryotic genes (Ropero et al., Citation2017). Mutation in this region may decrease the binding of some transcription factors; therefore, the promoters of the beta-genes will disappear in favor of the delta-genes, causing increases in the transcription of the delta-genes in patients with this type of β-thal and with the consequent increase in Hb A2 (Codrington et al., Citation1990). The (−29 A>G) mutation is common among blacks patients (Antonarakis et al., Citation1984), Chinese and Moroccan families (Huang et al., Citation1986; Lemsaddek et al., Citation2003). It is also recorded in Turkish and Spanish patients (Akar et al., Citation2003; Pereira Mdel, Dalmau, & Corrons, Citation2009). In surprising, this mutation was observed in a Syrian family originating from the central region of Syria, so this mutation maybe introduced to this region by population migrations.
In our case, the data Hb A1 = 12.5%, Hb F = 83.7%, and Hb A2 = 3.8% are compatible with the usual pattern of β+/β° type which shows Hb A = 10–30%, Hb F = 70–90%, and Hb A2 = 2–5% (Cao & Galanello, Citation2010). The fetal hemoglobin (Hb F) in patients with homozygous or compound heterozygous mutations in β-thal is in the range 70–90% (Cao & Galanello, Citation2010; Cao, Galanello, & Rosatelli, Citation1994). Hb F, formed by (2α+2γ) globin chains, is expressed at high levels during fetal development and its level decreases after birth (Cao & Galanello, Citation2010; Cao et al., Citation2011). In fact, the production of fetal Hb during adult life is very tightly regulated by several genetic factors, some of which are located near the γ-globin gene and others on different chromosomes (Weatherall & Clegg, Citation2001). In addition, the presence of the Xmn-I polymorphism that might be linked with increased level of Hb F and the co-inheritance of a globin gene deletions (Zalloua, Aoun, Koussa, Asfahani, & Taher, Citation2003), other study by Aditya et al. concluded that the presence of Xmn-I polymorphism and IVS-I-1 mutation leads to a milder phenotypic presentation causing a delay in onset of blood transfusions but does not affect the amount of blood received (Aditya, Verma, Saxena, Kaul, & Khanna, Citation2006). In our patient case, the level of Hb F was 83.7%, the type of mutation was compound heterozygous, and the genotype of Xmn-I polymorphism was homozygous. These factors may be contributed to ameliorate the form of β-thal intermedia (β-TI) phenotype. At 16 months of age, blood transfusion was done in our patient case, because he had a sudden decline in Hb levels caused by an upper respiratory infection. After the initial transfusion, he required frequent transfusions and recently, the splenomegaly is observed (15 cm).
5. Conclusion
We present here a case report of rare beta-globin mutation (−29 A>G) which was found in Syrian male patient. This mutation associated with (IVS-I-1 G>A) mutation and the rare genotype (−29 A>G/IVS-I-1 G>A) was found in Syrian patient. However, our finding suggests that the presence of this mutation in this region may be resulted from old migrations in Syria. This requires further substantiation with larger sample size.
Abbreviations
β-thal: Beta thalassemia
Hb: hemoglobin
IVS: Intervening sequence
Acknowledgements
The authors thank Prof. I. Othman, the Director General of Atomic Energy Commission of Syria (AECS), and Dr. N. Mirali, the head of Molecular Biology and Biotechnology Department, for their support. This work was supported by the AECS.
Additional information
Funding
Notes on contributors
Hossam Murad
Hossam Murad The research activity of our group focuses on studying the genetic diseases prevalent in Syria, especially the most common ones, by studying the mutations responsible of diseases, whether in a single gene or in a number of genes using molecular biological methods. We work in harmony with government hospitals and private clinics in order to obtain samples.
The study focused on a case of a child affected with beta-thalassemia, where we detected a rare genotype IVS-I-1 (G>A) (HBB:c.92+1 G>A)/−29 A>G (HBB:c.−79 A>G) on the β-globin gene causing β°/β+ thalassemia intermedia.
References
- Aditya, R., Verma, I. C., Saxena, R., Kaul, D., & Khanna, V. K. (2006). Relation of Xmn-1 polymorphism & five common Indian mutations of thalassaemia with phenotypic presentation in b-thalassaemia. JK Science : Journal of Medical Education and Research, 8(3), 139–143.
- Akar, E., Ozdemir, S., Timur, I. H., & Akar, N. (2003). First observation of homozygous hemoglobin hamadan (B 56 (D7) GLY-ARG) and beta thalassemia (−29 GA)-hemoglobin hamadan combination in a Turkish family. American Journal of Hematology, 74, 280–282. doi:10.1002/ajh.10404
- Antonarakis, S. E., Irkin, S. H., Cheng, T. C., Scott, A. F., Sexton, J. P., Trusko, S. P., … Kazazian, Jr, H. H. (1984). Beta-thalassemia in American blacks: Novel mutations in the “TATA” box and an acceptor splice site. Proceedings of the National Academy of Sciences of the United States of America, 81(4), 1154–1158. doi:10.1073/pnas.81.4.1154
- Beshara, A. (2011). The origins of Syrian nationhood: Histories, pioneers and identity. London and New York: Routledge, Taylor & Francis Group.
- Cao, A., & Galanello, R. (2010). Beta-thalassemia genereviews. Seattle, WA: Genet Med.
- Cao, A., Galanello, R., & Rosatelli, M. C. (1994). Genotype phenotype correlations in beta thalassemia. Blood Reviews, 8, 1–12. doi:10.1016/0268-960X(94)90002-7
- Cao, A., Moi, P., & Galanello, R. (2011). Recent advances in beta-thalassemias. Pediatric Reports, 3(2), 65–78. doi:10.4081/pr.2011.e17
- Codrington, J. F., Li, H. W., Kutlar, F., Gu, L. H., Ramachandran, M., & Huisman, T. H. (1990). Observations on the levels of Hb A2 in patients with different beta-thalassemia mutations and a delta chain variant. Blood, 76(6), 1246–1249.
- Giardine, B., Borg, J., Viennas, E., Pavlidis, C., Moradkhani, K., Joly, P., … Patrinos, G. P. (2014). Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Research, 42, D1063–1069. (Database). doi:10.1093/nar/gkt911
- Hardison, R. C., Chui, D. H. K., Giardine, B., Riemer, C., Patrinos, G. P., Anagnou, N., … Wajcman, H. (2002). HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutation, 19(3), 225–233. doi:10.1002/humu.10044
- Huang, S., Wong, C., Antonarakis, S. E., Ro-Lien, T., Lo, W. H., & Kazazian, H. H., Jr. (1986). The same “TATA” box beta-thalassemia mutation in Chinese and US blacks: Another example of independent origins of mutation. Human Genetics, 74(2), 162–164. doi:10.1007/BF00282081
- Indrak, K., Brabec, V., Indrakova, J., Chrobak, L., Sakalova, A., Jarosova, M., … GU, Y. C. (1992). Molecular characterization of beta-thalassemia in Czechoslovakia. Human Genetics, 88(4), 399–404.
- Jarjour, R. A., Murad, H., Moasses, F., & Al-Achkar, W. (2014). Molecular update of beta-thalassemia mutations in the Syrian population: Identification of rare beta-thalassemia mutations. Hemoglobin, 38(4), 272–276. doi:10.3109/03630269.2014.912661
- Lemsaddek, W., Picanco, I., Seuanes, F., Mahmal, L., Benchekroun, S., Khattab, M., … Osório-Almeida, L. (2003). Spectrum of beta thalassemia mutations and HbF levels in the heterozygous Moroccan population. American Journal of Hematology, 73(3), 161–168. doi:10.1002/ajh.10358
- Murad, H., Moassas, F., Jarjour, R., Mukhalalaty, Y., & Al-Achkar, W. (2014). Prenatal molecular diagnosis of beta-thalassemia and sickle cell anemia in the Syrian population. Hemoglobin, 38(6), 390–393. doi:10.3109/03630269.2014.978455
- Murad, H., Moasses, F., Dabboul, A., Mukhalalaty, Y., Bakoor, A. O., Al-Achkar, W., Jarjour, R. A. (2018). Geographical distribution of beta-globin gene mutations in Syria. Hematology, 23(9), 697–704. doi:10.1080/10245332.2018.1461291
- Pereira Mdel, M., Dalmau, A. C., & Corrons, J. L. (2009). Molecular heterogeneity of beta-thalassemia alleles in Spain and its importance in the diagnosis and prevention of beta-thalassemia major and sickle cell disorders. Hemoglobin, 33(3), 226–234. doi:10.1080/03630260903089003
- Ropero, P., Erquiaga, S., Arrizabalaga, B., Perez, G., de la Iglesia, S., Torrejon, M. J., … Martínez, R. (2017). Phenotype of mutations in the promoter region of the beta-globin gene. Journal of Clinical Pathology, 70(10), 847–848. doi:10.1136/jclinpath-2017-204378
- Rujito, L., Basalamah, M., Siswandari, W., Setyono, J., Wulandari, G., Mulatsih, S., … Sutaryo, S. (2016). Modifying effect of XmnI, BCL11A, and HBS1L-MYB on clinical appearances: A study on beta-thalassemia and hemoglobin E/beta-thalassemia patients in Indonesia. Hematology/Oncology and Stem Cell Therapy, 9(2), 55–63. doi:10.1016/j.hemonc.2016.02.003
- Tadmouri, G. O., & Gulen, R. I. (2003). Deniz: The electronic database for beta-thalassemia mutations in the Arab world. Saudi Medical Journal, 24(11), 1192–1198.
- Thein, S. L. (2013). The molecular basis of beta-thalassemia. Cold Spring Harbor Perspectives in Medicine, 3(5), a011700. doi:10.1101/cshperspect.a011700
- Villegas, A., Ropero, P., Gonzalez, F. A., Anguita, E., & Espinos, D. (2001). The thalassemia syndromes: Molecular characterization in the Spanish population. Hemoglobin, 25(3), 273–283. doi:10.1081/HEM-100105220
- Weatherall, D. J., & Clegg, J. B. (2001). Inherited haemoglobin disorders: An increasing global health problem. Bulletin of the World Health Organization, 79(8), 704–712.
- Yassin, M. M., Sirdah, M. M., Al Haddad, R. M., Lubbad, A. H., & Al-Yazji, M. S. (2013). Genotype-phenotype characteristics of β thalassemia children in the Gaza Strip, Palestine. Journal of Genetic Disorders & Genetic Reports, 02(02), 1–6. doi:10.4172/2327-5790.1000109
- Zalloua, P. A., Aoun, E., Koussa, S., Asfahani, W. S., & Taher, A. (2003). The codons 8/9 (+G) mutation found for the first time in the Lebanese population. Hemoglobin, 27(1), 1–5. doi:10.1081/HEM-120018430