
ABSTRACT
We report the therapeutic potential of GSK621, an AMP-activated protein kinase (AMPK) agonist, in acute myeloid leukemia (AML). GSK621-induced cytotoxicity is restricted to AML cells compared to normal hematopoietic progenitors due to a unique synthetic lethal interaction of co-activation of AMPK and mammalian target of rapamycin complex 1 (mTORC1) that involves the stress response pathway. AMPK activation thus represents an attractive perspective for cancer therapy.
KEYWORDS:
Acute myeloid leukemia (AML) arises from the transformation of hematopoietic progenitor cells, which results in excessive proliferation of immature cells leading to bone marrow failure. Current treatments involve intensive chemotherapy and hematopoietic stem cell transplantation in young patients. However, in elderly patients the treatment options are limited and palliative care is usually given, underscoring the need for new treatment modalities for this disease.
AML displays heterogeneous genetic and phenotypic features that present a challenge to the development of targeted therapies. However, regardless of their genetic background, AML cells show activation of common oncogenic signaling pathways that represent potential targets for new therapies. Many experimental findings pinpoint mammalian target of rapamycin complex 1 (mTORC1) as a target for drug development in cancer because of its central role in promoting anabolism through protein translation or nucleotide biosynthesis activation.Citation2 mTORC1 is constitutively overactivated in AML cells, but not in normal hematopoietic progenitors, although the molecular mechanism involved in this abnormal activity has remained elusive. Thus far, treatment of AML patients with first-generation mTORC1 inhibitors (i.e., rapamycin and its derivatives) has turned out to be disappointing.
In an attempt to more efficiently block mTORC1 activity for clinical applications in AML patients, we and other groups have used metformin as a means to activate AMP-activated kinase (AMPK).Citation3 AMPK is a heterotrimeric complex that is activated in the context of energy stress by the fixation of AMP to its γ-catalytic subunit and by phosphorylation of its α-catalytic subunit involving liver kinase B1 (LKB1).Citation4 Upon activation, AMPK turns down anabolism through inhibition of acetyl-CoA carboxylase (ACC), a key enzyme in fatty acid synthesis, or mTORC1. AMPK also promotes catabolism, including fatty acid oxidation, or autophagy to restore cellular energy balance.Citation4 Inhibition of mitochondrial oxidative phosphorylation in AML cells by metformin activates AMPK and reduces AML propagation in vitro and in vivo in mice models.Citation3 However, Scotland and colleagues have shown that in AML, as in other cancer types,Citation5 the cytotoxic effects of metformin are mostly AMPK-independent, leaving unanswered the question of whether AMPK activation may represent a therapeutic modality in cancer.
In a recent study, we used GSK621, a new thienopyridone-derived molecule, to activate AMPK in AML cells. Using clustered regularly interspaced short palindromic repeats (CRISPR) or RNA interference, we inactivated AMPK and observed that AMPKα1-depleted AML cells were protected from GSK621-induced cytotoxicity, demonstrating the specificity of this new compound against its target at the cellular scale. When assayed in vitro against 20 AML cell lines and 16 primary samples from AML patients harboring a broad range of genetic alterations including poor-prognostic mutations such as Fms-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD), GSK621 induced cytotoxicity with a median IC50 of 20 µM. Treatment with GSK621 also reduced the stemness of oncogene-transformed mouse hematopoietic progenitors in repeated clonogenic assays in vitro, suggesting that AMPK activation may deplete the most immature cellular leukemic population.
From a therapeutic perspective, differences in GSK621-induced cytotoxicity between AML and normal hematopoietic cells attracted our attention. We first hypothesized that AMPK activation might disrupt the oncogenic addiction of AML cells to mTORC1, which is absent in normal hematopoietic progenitor cellsCitation6 (). Unexpectedly, we found that mTORC1 activity was preserved in GSK621-treated AML cells even if AMPK was fully activated as attested by biochemical readouts such as ACC phosphorylation. Strikingly, opposite results were seen in normal hematopoietic progenitors, in which GSK621 treatment ablated mTORC1 activity. This dissociated AMPK-mTORC1 axis was also recently observed in glioma cells.Citation7 The different behavior of transformed versus non-transformed hematopoietic cells supports AMPK activation as a promising therapeutic modality in AML.
Figure 1. Oncogenic addiction versus synthetic lethality to explain GSK621-induced cytotoxicity in AML. (A) In the oncogenic addiction model, GSK621 activates AMPK. Upon activation, AMPK inactivates mTORC1 (mTOR/raptor complex) by direct phosphorylation of raptor. AMPK also indirectly inhibits mTORC1 via an activating phosphorylation of TSC1/2 (Tuberous Sclerosis Complex ½) harboring a GAP (GTPase activating protein) activity toward rheb (ras-homolog enriched in brain), a small G-protein involved in mTORC1 activation. Hence, AMPK-dependent TSC1/2 activation favors inactive GDP-bound rheb, ultimately inhibiting mTORC1 activity. (B) In the synthetic lethality model, AMPK and mTORC1 activities are uncoupled. In AML cells, mTORC1 is overactivated due to unidentified upstream signals. Co-activation of AMPK and mTORC1 activates the eIF2α signaling pathway, which triggers autophagic cell death accounting for GSK621-induced cytotoxicity. In addition, eIF2α activation and autophagy might stimulate cancer immunogenicity through dendritic cell-mediated activation of cytotoxic T lymphocytes.
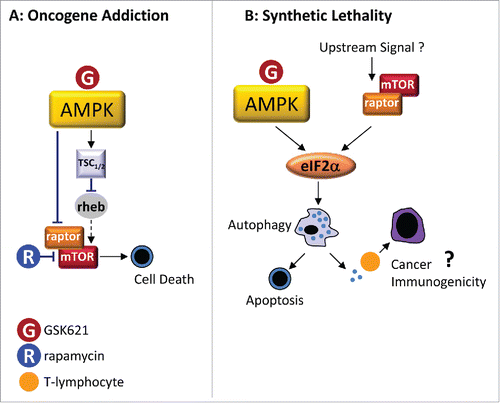
To integrate our new findings, we proposed an alternative hypothesis: GSK621 may be selectively toxic to AML cells as a result of the sustained mTORC1 activity upon AMPK activation (). Consistent with this hypothesis, we observed that mTORC1 inhibition by pharmacologic (using rapamycin) or genetic means protected AML cells from cytotoxicity induced by AMPK activators (GSK621 and also A769662 and 991, 2 other direct and specific AMPK agonistsCitation1). To further confirm our hypothesis, we forced mTORC1 activity in AML cells (CRISPR-induced TSC2 [Tuberous Sclerosis Complex 2] deletion) and normal hematopoietic progenitors (expression of a constitutively activated AKT allele) and observed an increased sensitivity to GSK621 that was abrogated by rapamycin. Our results were thus consistent with a synthetic lethal interaction of AMPK and mTORC1 co-activation. The concept of synthetic lethality came from genetics: 2 genes are synthetic lethal if mutation of either alone is compatible with cell viability but concomitant mutation induces cellular death.Citation8 Building on this concept, 2 hits against cancer cells are synthetic lethal when they do not affect cell viability separately—as is the case for activation of AMPK and mTORC1 in AML cells—but synergize to kill cancer cells.
From a molecular perspective, we linked this synthetic lethal interaction to the stress response pathway. GSK621 activated the translation-initiating factor 2α (eiF2α), which controls protein translation independent of mTORC1 and promotes autophagy and apoptosis through the control of transcription factors such as activating transcription factor 4 (ATF4). Pharmacologic or genetic attenuation of eIF2α-dependent effectors reduced GSK621-induced cytotoxicity and rapamycin prevented GSK621-dependent eIF2α activation. The notion that treatment with AMPK agonists may induce autophagy has therapeutic implications. We showed that GSK621-induced autophagy was not protective in our model—in contrast to most models—but was involved in autophagic cell death that accounted for a significant proportion of GSK621-induced cytotoxicity. Besides this cell-intrinsic effect, triggering eIF2α may be relevant for cancer immunogenicity due to the release of mediators stimulating specific tumor-targeting adaptive immunity.Citation9 This perspective is particularly exciting with regard to the recent development of immunomodulatory drugs targeting programmed cell death 1/programmed cell death 1 ligand (PD-1/PDL-1) that have shown impressive activity across various cancer cell types.Citation10
Together, our findings suggest that targeting AMPK activation may be a valuable therapeutic strategy in mTORC1-overactivated cancers. Identification of other pathways that offer synthetic lethal hits with AMPK activation or agents with potential synergy with AMPK agonists—such as immune checkpoint blockade drugs—represents a fascinating challenge for personalized cancer medicine.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The study described here was made possible by the sustained efforts of the following scientists: Poulain Laury, Paubelle Etienne, Zylbersztejn Florence, Grenier Adrien, Lambert Mireille, Townsend Elizabeth C., Brusq Jean-Marie, Nicodeme Edwige, Decroocq Justine, Nepstad Ina, Green Alexa S., Mondesir Johanna, Hospital Marie-Anne, Jacque Nathalie, Christodoulou Alexandra, Desouza Tiffany A., Hermine Olivier, Foretz Marc, Viollet Benoit, Lacombe Catherine, Mayeux Patrick, Weinstock David M., Moura Ivan C. and Bouscary Didier.
Funding
Pierre Sujobert was supported by grants from the French INSERM (Institut National de la santé et de la recherché médicale) Institute and from the French ARC (Association pour la Recherche sur le Cancer) association. This work was supported by institutional funding from INSERM and by grants from LNCC (Ligue Nationale Contre le Cancer, équipe labélisée) and from INCa (Institut National du Cancer).
References
- Sujobert P, Poulain L, Paubelle E, Zylbersztejn F, Grenier A, Lambert M, Townsend EC, Brusq JM, Nicodeme E, Decrooqc J, et al. Co-activation of AMPK and mTORC1 induces cytotoxicity in acute myeloid leukemia. Cell Rep 2015; 11:1446-57; PMID:26004183; http://dx.doi.org/10.1016/j.celrep.2015.04.063
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, Boyer O, Bardet V, Park S, Foretz M, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood 2010; 116:4262-73; PMID:20668229; http://dx.doi.org/10.1182/blood-2010-02-269837
- Hardie DG. The LKB1-AMPK pathway-friend or foe in cancer? Cancer Cell 2013; 23:131-2; PMID:23410967; http://dx.doi.org/10.1016/j.ccr.2013.01.009
- Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, Senegas I, Peyraud R, Peyriga L, Theodoro F, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 2013; 27:2129-38; PMID:23568147; http://dx.doi.org/10.1038/leu.2013.107
- Recher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, Mas VM, Benzaquen D, Laurent G, Huguet F, Payrastre B. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 2005; 105:2527-34; PMID:15550488; http://dx.doi.org/10.1182/blood-2004-06-2494
- Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, Kumar A, Zhou X, Sun Y, Quinn B, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A 2014; 111:E435-44; PMID:24474794; http://dx.doi.org/10.1073/pnas.1311121111
- Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5:689-98; PMID:16110319; http://dx.doi.org/10.1038/nrc1691
- van Vliet AR, Martin S, Garg AD, Agostinis P. The PERKs of damage-associated molecular patterns mediating cancer immunogenicity: from sensor to the plasma membrane and beyond. Semin Cancer Biol 2015; PMID:25882379
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. New Engl J Med 2015; 372:311-9; PMID:25482239; http://dx.doi.org/10.1056/NEJMoa1411087