
ABSTRACT
Our recent study shows that AMPK normally phosphorates fumarase (FH) at Ser75 under glucose deprivation, resulting in FH-ATF2 complex formation that facilitates transcription for cell growth arrest. Meanwhile, O-GlcNAc transferase can compete with AMPK to O-GlcNAcylate FH. In tumor cells, FH is highly O-GlcNAcylated and is proinhibited from AMPK-ATF2 signaling.
Tumor cells always confront pressure of nutrient shortage due to their rapid nutrient consumption and limited supply from surrounding vascular, while they still keep the sustainable growth.Citation1 It is well known that tumor cells can alternatively take strategies such as autophagy or macropinocytosis to conquer nutrient-crisis in terms of anabolic requirement.Citation2 Besides, the mechanisms that how to effectively resist nutrient deficiency-induced stress signaling is also indispensable for tumor cells to facilitate the survival,Citation3 as the detrimental effects under this condition would be brought out more fiercely than that from the defect of building blocks-maintenance. Hence, to understand the tumor cell-addicted mechanisms underlying counteractive responses to nutrient stress can be crucial for developing effective treatment in cancer therapy.
Fumarase (FH), a key enzyme involved in tricarboxylic acid cycle, catalyzes the reversible hydration and dehydration reaction of fumarate to malate.Citation4 The previous study demonstrates FH is accumulated at DNA breaks area after DNA damage, and vitally participates in DNA repair through its local fumarate production.Citation5 The regulation of DNA repair by FH is attributed to the inhibitory effects of fumarate on α-KG-dependent histone demethylases, which soundly affects histone methylation required for the progression of DNA repair.Citation5 Given the general role of histone methylation linking to diverse cellular activities,Citation6 these findings inspire us further to investigate whether the path of FH-histone methylation can modulate gene transcription under certain physiologic context such as nutrient deficiency.
O-GlcNAcylation is an important protein modification at ser/thr residues and is reversely catalyzed by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA).Citation7 Protein O-GlcNAcylation is tightly regulated by glucose availability, implying its relevance to cellular metabolic status.Citation8 High OGT activity has been reported to be prevalently implicated in tumor progression,Citation9 although the concrete mechanisms remain to be further clarified.
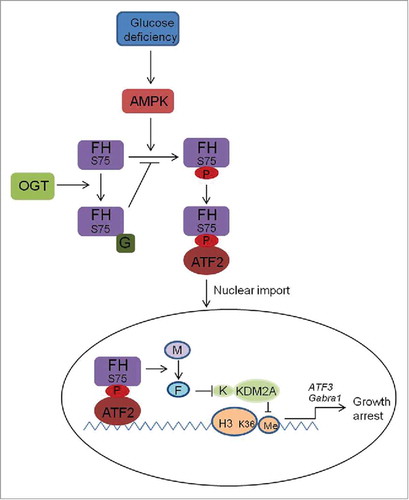
In our study,Citation10 it was first investigated whether FH is involved in cellular signaling transduction in response to glucose deficiency. Mass spectrum followed by immunoprecipitaion analysis in normal human pancreatic duct epithelial (HPDE) cells indicates FH is specifically associated with ATF2 (an AP-1 transcription factor family member) under glucose deprivation. In line with this, ChIP analysis shows glucose deprivation readily induces FH recruitment to the promoters of ATF2-targeted genes such as ATF3 and C-JUN. Subsequent biochemical analyses indicate FH-ATF2 interaction is dependent on AMPK activation and AMPK can directly phosphorate FH at Ser75. Importantly, FH S75A mutant largely blocks its interaction with ATF2 and the physiologic events under glucose deprivation. Given the important characteristics of FH as a metabolic enzyme, catalytically inactive FH R233H is used, and we found that glucose deprivation-induced cell proliferation arrest is blocked by expression of FH R233H, although it remains similar ATF2 binding ability to WT FH. Additionally, high concentration of exogenous fumarate but not malate partially reverses the repression effect of FH S75A and FH R233H on transcription and cell growth arrest. Meanwhile, our epigenetic analysis further indicates promoter-associated FH inhibits α-KG-dependent histone demethylase KDM2A activity through the local fumarate production, which maintains H3K36me2 levels for expression of genes relevant to cell cycle arrest. Thus, these results suggest that either local promoter association or metabolic activity of FH is crucial for ATF2-mediated downstream effects.
In contrast with normal cells, FH-ATF2 interaction is only weakly detected in pancreatic cancer cells under glucose deficiency. Along with the results that FH-ATF2 interaction is exclusively induced by glucose but not glutamine deprivation, we assumed that protein O-GlcNAcylation, which is closely associated with glucose availability, would be importantly involved. Pancreatic cancer cell lines display the significantly higher OGT levels compared with normal cells. The results from OGT overexpression or depletion suggest that FH-ATF2 interaction was inversely related to O-GlcNAcylation. Intriguingly, in vitro O-GlcNAcylation analysis indicates that FH could be O-GlcNAcylated at the AMPK phosphorylation site Ser75, which is greatly supported by the results of mass spectrum analysis showing FH Ser75 underwent O-GlcNAcylation. These data provide the direct evidence for the negative regulation of FH-ATF2 interaction by O-GlcNAcylation. Functionally, cells in vitro assay and xonograft cancer model in nude mice both demonstrate FH O-GlcNAcylation prevented FH-ATF2-mediated suppression of tumor cell growth. Clinically, Analysis of human tumor samples reveals the inverse correlation between FH-Ser75 phosphorylation and OGT activity and poor prognosis in patients.
In brief, our study shows that in glucose-deprived normal pancreatic cells, AMPK phosphorylates FH at Ser75 and promotes its complex formation with ATF2 at the targeted genes promoters. Promoter-associated fumarase exhibits enzymatic effect and inhibits KDM2A–mediated H3K36me2 demethylation, thereby results in H3K36me2-dependent gene expression for cell growth arrest. Secondly, OGT is found to O-GlcNAcylate FH at AMPK phosphorylation site, and this effect is amplified in cancer cells displaying strong OGT activity, which impedes AMPK-FH-ATF2 signaling and maintains tumor cell growth under glucose deficiency ().
Figure 1. Transcription regulation by FH under the competitive modifications mediated by AMPK and OGT: AMPK phosphorates FH at Ser75 under glucose deficiency and induces FH-ATF2 complex formation and subsequent promoter association to activate cell cycle arrest gene expression. OGT competes with AMPK to O-GlcNAcylate FH at Ser75 and inhibits AMPK-FH-ATF2-mediated downstream effects. G, O-GlcNAcylation; M, malate; F, fumarate; K, α-KG; Me, methylation.
Collectively, these findings on one hand reveal an unidentified mechanism underlying the subtle regulation of gene transcription by metabolic enzymes, and on the other hand demonstrate that cancer cells bias OGT-mediated effects to overcome stress signaling under nutrient deficiency, of which the molecular basis potentially provides critical clues for improving therapies against tumors with high OGT activity.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364-73. doi:10.1038/nature11706. PMID:23151579
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361-5. doi:10.1038/nature14587. PMID:26168401
- Efeyan A, Comb WC. Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517,:302-10. doi:10.1038/nature14190. PMID:25592535
- Kornberg H.L., Krebs HA. Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature. 1957;179:988-91. doi:10.1038/179988a0. PMID:13430766
- Jiang Y, Qian X, Shen J, Wang Y, Li X, Liu R, Xia Y, Chen Q, Peng G, Lin SY, et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nature cell biology. 2015;17:1158-68. doi:10.1038/ncb3209. PMID:26237645
- Greer E.L, Shi Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nature reviews Genetics. 2012;13:343-57. doi:10.1038/nrg3173. PMID:22473383
- Nagel AK, Ball LE. O-GlcNAc transferase and O-GlcNAcase: Achieving target substrate specificity. Amino acids. 2014;46:2305-16. doi:10.1007/s00726-014-1827-7. PMID:25173736
- Hardiville S. & Hart GW. Nutrient regulation of gene expression by O-GlcNAcylation of chromatin. Current opinion in chemical biology. 2016;33:88-94. doi:10.1016/j.cbpa.2016.06.005. PMID:27322399
- Ferrer C.M, Reginato MJ. Sweet connections: O-GlcNAcylation links cancer cell metabolism and survival. 2015;2:e961809
- Wang T, Yu Q, Li J, Hu B, Zhao Q, Ma C, Huang W, Zhuo L, Fang H, Liao L, et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. 2017;19:833-43