
ABSTRACT
Metastatic melanoma remains incurable for many due to its aggressive nature. Secreted cathepsins promote metastasis by cleaving matrix and activating pro-invasive proteases. We reported that ABL kinases induce cathepsin secretion and subsequent metastasis by activating ETS1, SP1, and RELA pathways, indicating that ABL inhibitors may serve as novel anti-cathepsin agents.
Melanoma is highly aggressive, and despite recent new therapeutic advances, many patients with metastatic melanoma still succumb to the disease (5-year survival rate-18%; https://seer.cancer.gov/data/citation.html.) Cysteine cathepsin proteases, whose expression and secretion is dramatically increased in cancer, have well-recognized roles during cancer progression.Citation1 In cancer cells, cathepsin protein and mRNA is often dramatically increased, and excess procathepsins (unprocessed proforms) are exocytosed and secreted.Citation1 Extracellular cysteine cathepsins have been isolated from the media from cancer cells where they comprise >40% of total secreted proteins, and are observed in the serum from cancer patients.Citation2 Secreted cathepsins promote melanoma invasion and metastasis by directly and indirectly promoting degradation of the extracellular matrix.Citation1 Thus, due to their dysregulation during cancer progression and association with therapeutic resistance, cathepsins are attractive targets.Citation1 However, few inhibitors targeting cysteine cathepsins have reached the clinic due to issues with toxicity and lack of efficacy.Citation2 Thus, other strategies aimed at inhibiting cathepsin secretion may be more successful as anti-cancer therapies.
ABL family non-receptor tyrosine kinases (ABL1, ABL2-also known as Arg) are most known for their involvement in human leukemia.Citation3 However, we found that the kinases also are activated in melanoma cell lines and primary tumors, downstream of receptor tyrosine kinases, SRC Family Kinases (SFKs) and BRAF/ERK signaling, and promote melanoma development and progression.Citation4-6 In our recent findings published in “Science Signaling”, we used loss-of-function (si/shRNA; ABL1/ABL2 inhibitors, nilotinib and GNF-5) and gain-of-function approaches to demonstrate that ABL1 and ABL2 promote CTSB and CTSL1 (a.k.a. cathepsin B, cathepsin L) expression and subsequent secretion from melanoma cells by promoting CTSB and CTSL1 mRNA expression and promoter activity. Our findings in cell lines also were extended to patient samples as ABL1/ABL2 and CTSL1/CTSB mRNAs were increased in melanoma samples from patients, and were correlated in those samples.Citation7
We next identified the mechanism by which ABL1 and ABL2 induce cathepsin expression. We found that ABL1/ABL2 increased the abundance, phosphorylation, nuclear translocation, and/or promoter activity of several transcription factors with known roles in melanoma progression (SP1, ETS1, RELA), and activation of these transcription factors subsequently induced cathepsin expression and secretion. Importantly, we also found that ABL1 and ABL2 dramatically induced the DNA-binding activity of SP1 to CTSB and CTSL1 promoters; ETS1 to the CTSL1 promoter; and RELA to the CTSB promoter (). Moreover, we demonstrated clinical relevance by showing that ABL1/ABL2 and ETS1/SP1 mRNAs were correlated in primary melanomas.Citation7
Figure 1. ABL kinase regulation of cathepsin secretion in melanoma. ABL1/ABL2 are activated by receptor tyrosine kinases (RTKs), SRC Family Kinases (SFK) and BRAF-ERK, and subsequently induce SP1, ETS1, and RELA expression/nuclear translocation, and binding to cathepsin promoters. These events induce cathepsin transcription, secretion, invasion, and metastasis. PM: Plasma Membrane; ER: Endoplasmic Reticulum; and ECM: Extracellular Matrix.
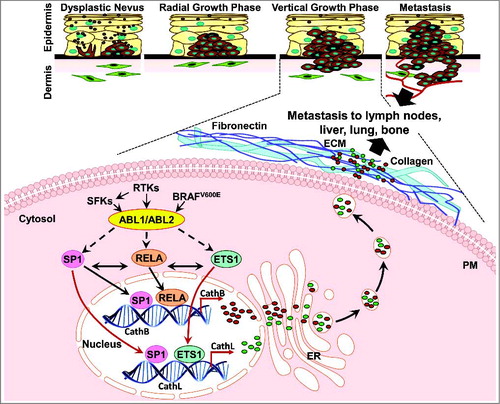
Interestingly, the signaling pathways we identified showed cell-type specific expression. ABL1/ABL2 induced cathepsin expression and secretion via a mechanism requiring their kinase activities in most melanoma lines examined (e.g. WM3248, LOX-IVMI, UACC-903); however, in 435s cells and BT-549 breast cancer cells, ABL1/ABL2 promoted cathepsin secretion via a kinase-independent mechanism. Kinase-inactive forms of ABL1 can retain scaffold/adapter-like signaling functions, likely mediated by binding proteins or protein complexes to one of their signaling domains (e.g. SH3, SH2, polyproline motif);Citation3 however, this type of signaling had not previously been documented in melanoma. The kinase-dependent mechanism by which ABL1 and ABL2 induce cathepsin expression/secretion involved cooperative and bidirectional regulation of SP1, ETS1, and RELA transcription factors. In contrast, the kinase-independent pathway identified in 435s cells did not involve SP1 but rather, ABL1/ABL2 induced cathepsin expression/secretion via ETS1 and RELA. We also identified RELA as the convergent point in both pathways as it mediated ABL1/ABL2-dependent induction of cathepsin secretion in both pathways.
Importantly, the pathways we identified also have functional, biological, and in vivo relevance. Silencing or inhibiting (using nilotinib) ABL1/ABL2 dramatically blocked melanoma matrigel invasion and metastatic lung colonization (intravasation/extravasation) in a mouse model, and these effects were efficiently rescued by forced expression/secretion of cysteine cathepsins.Citation7 Moreover, rescue of ABL1/ABL2 siRNA- or nilotinib-mediated inhibition of invasion was prevented by incubation with E64C, a cysteine cathepsin inhibitor that is impermeable to the cell membrane, which indicates that extracellular cathepsins mediate the rescue. These data thus indicate that ABL1/ABL2 drive melanoma invasion and metastasis, in large part, by inducing cathepsin secretion.
Increased cathepsin expression and subsequent secretion are critically important for invasion and metastasis of melanoma cells; however, the signaling pathways driving this process have not been extensively studied. In our “Science Signaling” report, we demonstrate that activated ABL1 and ABL2 are responsible for inducing cathepsin mRNA, protein, and secretion and do so by influencing the nuclear expression and DNA binding activity of RELA, ETS1, and SP1. Although the data shown were focused on cathepsins B/L, ABL1/ABL2 also regulate other cysteine cathepsins that are linked to melanoma dissemination (K,S), which indicates that ABL1/ABL2 may regulate melanoma metastasis by inducing secretion of a variety of pro-invasive cathepsins perhaps via similar pathways.Citation7 Our findings also extend beyond induction of cathepsins as ETS1, SP1 and RELA regulate numerous targets that drive processes crucial for cancer progression including epithelial-mesenchymal-transition (EMT), regulation of stem cell traits, metabolic reprogramming, and drug resistance (including to immunotherapy), and their overabundance is associated with a poor prognosis.Citation8-10 Despite their crucial roles in cancer progression, transcription factors are notoriously hard to target, and few drugs targeting cathepsins have reached the clinic. Our data demonstrate that targeting ABL1/ABL2 is a new way not only to prevent cathepsin secretion, but also to reduce the expression and activity of ETS1, SP1, and RELA. Since FDA-approved drugs targeting ABL1/ABL2 have been used for decades to treat leukemia, with relatively few side-effects, these data have strong translational significance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
The Research Communications Office of the Markey and Center (P30CA177558) supported this work and contributed to the figure.
Additional information
Funding
References
- Olson OC, Joyce JA. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat Rev Cancer. 2015;15(12): 712–29.
- Sudhan DR, Siemann DW. Cathepsin L targeting in cancer treatment. Pharmacol Ther. 2015;155:105–16.
- Ganguly SS, Plattner R. Activation of Abl family kinases in solid tumors. Genes Cancer. 2012;3(5-6):414–25.
- Fiore LS, Ganguly S, Sledziona J, Cibull ML, Wang C, Richards DL, Neltner JM, Beach C, McCorkle JR, Kaetzel DM, et al. c-Abl and Arg induce cathepsin-mediated lysosomal degradation of the NM23-H1 metastasis suppressor in invasive cancer. Oncogene. 2014;33(36):4508–20.
- Ganguly SS, Fiore LS, Sims JT, Friend JW, Srinivasan D, Thacker MA, et al. c-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene. 2012;31(14):1804–16.
- Jain A, Tripathi R, Turpin CP, Wang C, Plattner R. Abl kinase regulation by BRAF/ERK and cooperation with Akt in melanoma. Oncogene. 2017;36(32):4585–96.
- Tripathi R, Fiore LS, Richards DL, Yang Y, Liu J, Wang C, Plattner R. Abl and Arg mediate cysteine cathepsin secretion to facilitate melanoma invasion and metastasis. Sci Signal. 2018;11(518).
- Dittmer J. The role of the transcription factor Ets1 in carcinoma. Semin Cancer Biol. 2015;35:20–38.
- Murtas D, Piras F, Minerba L, Ugalde J, Piga M, Maxia C, Perra MT, Sirigu P. Nuclear factor-kappaB expression is predictive of overall survival in patients with cutaneous melanoma. Oncol Lett. 2010; 1(4): 633–9.
- Vizcaino C, Mansilla S, Portugal J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol Ther. 2015;152:111–24.