
Ewing sarcoma is an aggressive pediatric cancer that is driven by a fusion oncogene, EWS-FLI1. While the transcriptional targets of EWS-FLI1 have been widely studied, the systemic impact of EWS-FLI1 transcription is relatively less understood. Here, we comment on our recent findings, published in Nature, regarding EWS-FLI1 driven transcription dysregulation and its impact on DNA damage response, BRCA1 function and R-loop accumulation.
Ewing sarcoma is the prototypical example of a solid tumor that is driven by chimeric oncogenes derived from chromosomal translocation. In over 85% of patients, the translocation is between chromosomes 22 and 11, fusing the N-terminal transactivation domain of the EWS RNA binding protein 1 (EWSR1) gene to the C-terminal DNA binding domain of the weak transcription factor Fli-1 proto-oncogene, ETS transcription factor (FLI1) gene.Citation1 The resultant EWS-FLI1 oncogene is a highly expressed transcription factor that modulates both gene and protein-regulatory networks to drive tumor initiation and maintenance. Despite the presence of this unique genetic event, conventional chemotherapy remains the treatment of choice for Ewing sarcoma. While effective in a significant number of cases, the long-term ramifications of the aggressive treatment regime necessitate understanding the basis of chemosensitivity and identifying alternative therapeutic targets.Citation2 We recently published in NatureCitation3 that EWS-FLI1 drives increased global transcription to cause a widespread accumulation of R-loops. This provides an obvious explanation to the increased replication stress that we and others have observed. However, we also chanced upon some unusual observations that provide insight into the PARP1 inhibitor sensitivity and mechanisms of chemoresistance.
A new class of BRCAness tumors
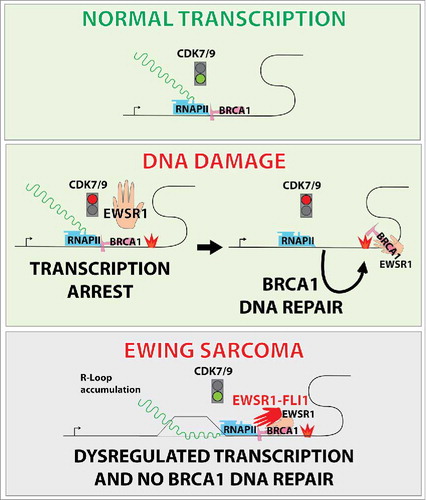
DNA damage induces profound changes to the transcriptional landscape, with both a targeted transcriptional program and a global shut down of general transcription.Citation4 We provide evidence of an active link between transcription stress and recombinational repair; EWSR1 dependent recruitment of the BRCA1, DNA repair associated (BRCA1) protein from transcription complexes to sites of damage is necessary for efficient homologous recombination. We speculate that in Ewing sarcoma, BRCA1 is retained not only at canonical target gene loci but also in EWS-FLI1 driven genes, further depleting BRCA1 availability. However, the mechanism by which this occurs remains an open question. Work in the 1990s demonstrated that BRCA1 is associated with the active transcription complexCitation5 and was later shown to release from this complex following DNA damage. Without EWSR1, or in the presence of EWS-FLI1, damage-induced global transcription shut down does not occur, and BRCA1 cannot relocate to sites of damage.
BRCA1 retention with active transcription complexes in Ewing sarcoma precludes its participation in homologous recombination, thereby explaining the acute sensitivity to poly(ADP-ribose) polymerase 1 (PARP1) inhibitors.Citation6 Therefore, this study also reveals a new class of BRCA1 deficient tumors that could benefit from PARP1 inhibitor treatments. However, chemoresistance mechanisms that arise in BRCA1 deficient carcinomas and circumvent the need for BRCA1 promoting recombination would also likely confer both chemotherapy and PARP1 inhibitor resistance in Ewing sarcoma. In fact, depletion of tumor protein p53 binding protein 1 (TP53BP1) both restores homologous recombination despite the presence of EWS-FLI1, and confers chemotherapy resistance to Ewing sarcoma cells. These observations may explain the lack of response observed in single agent PARP1 inhibitor clinical trials which were conducted with relapsed, chemoresistant patients.Citation7
EWS-FLI1: Dominant-negative over EWSR1
The EWS-FLI1 fusion protein is known to drive an oncogenic transcriptome, but here we demonstrate that it also interferes with EWSR1 function, thereby contributing to Ewing sarcoma pathogenesis. The most extensive knowledge of EWSR1 function stems from protein interaction studies that suggest a bridging function between transcription, RNA metabolism and genome surveillance.Citation8 There are mixed opinions in the field regarding its proto-oncogenic versus tumor suppressor role. We observed that EWSR1 inhibits the activation of RNA Polymerase II whereas EWS-FLI1 promotes the same, even in the presence of wild type EWSR1. Biochemical data indicated this was via the regulation of cyclin dependent kinase 9 (CDK9) (). These findings indicate a potential dominant-negative function of EWS-FLI1 over EWSR1. We also report that EWSR1 inhibits R-loop accumulation and facilitates homologous recombination. EWSR1 may be important for the release of BRCA1 from those transcribed genes with which it associates, and the concerted involvement of both proteins is essential for efficient recombination upon damage.
Figure 1. Altered control of transcription in Ewing sarcoma. Transcription elongation is triggered by cyclin dependent kinase 9 (CDK9) dependent phosphorylation of RNA Polymerase II (RNAPII) C-terminal domain. EWS RNA binding protein 1 (EWSR1) inhibits this phosphorylation event thereby controlling transcription rates. BRCA1, DNA repair associated protein (BRCA1) is associated with hyper-phosphorylated RNAPII under basal conditions but is released upon infliction of DNA damage to sites of break repair. However in Ewing sarcoma, EWS-FLI1 allows aberrant transcription by lifting EWSR1 dependent inhibition of CDK9 activity resulting in R-loop accumulation. This, in turn, prevents BRCA1 release from transcription complexes thus impairing homologous recombination repair of exogenously induced double strand breaks.
Lack of genome instability
One paradoxical observation from our work is that R-loops are thought to promote mutagenesis,Citation9 but genomic instability is largely absent in Ewing sarcoma,Citation10 with an average of five mutations found per primary tumor beyond the translocation. This is further compounded by the presence of high levels of replication stress and the lack of homologous recombination. In fact, the high levels of transcriptional stress, replication stress and the lack of homologous recombination conferred by EWS-FLI1 explain why expression of this oncogene is highly toxic to almost every cell type. This leads to a key question in the field regarding the cell-of-origin of Ewing sarcoma. It will be interesting to ask if neural crest stem cells or bone marrow mesenchymal stem cells, the putative cells of origin, provide a special molecular context that is permissive to the increased R-loops, replication stress and homologous recombination deficiency. Irrespective, these findings raise significant questions about the consequences of these defects, the associated genome instability and whether genomic instability in of itself is important for cancer development in all instances, or a secondary consequence in some cases. It is also important to note that genomes of tumors that survive chemotherapy are not stable. This suggests a “ceiling-effect” for the damage tolerance of Ewing sarcoma.
In conclusion, the present study highlights the contribution of EWSR1 loss to tumorigenesis and the unexpected outcomes of elevated R-loops in Ewing sarcoma. Pediatric cancers have comparatively stable backgrounds. This gives us the opportunity to study the biological processes that contribute to tumorigenesis or present therapeutic targets in relative isolation. We believe that Ewing sarcoma stands as an ideal system to study key processes involved in cancer biology without confounding mutations. Overall this work raises more questions than answers, and we hope that our findings will lead to asking the right questions to better understand this disease as well as identifying novel treatment strategies.
Financial disclosures
None
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by funds from NIH (K22ES012264, 1R15ES019128, 1R01CA152063), Voelcker Fund Young Investigator Award and CPRIT (RP150445) to A.J.R.B.; CPRIT (RP101491), Translational Science Training Across Disciplines Scholarship (UTHSA) and NCI postdoctoral training grant (T32CA148724) to A.G.
Additional information
Funding
References
- Delattre O. Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165.
- Rodriguez-Galindo C, Spunt SL, Pappo AS. Treatment of Ewing sarcoma family of tumors: Current status and outlook for the future. Med Pediatric Oncol. 2003;40:276–287.
- Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, Iniguez AB, Bernard X, Masamsetti VP, Roston S, et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature. 2018;555:387–391.
- Ljungman M, Zhang F. Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene. 1996;13:823–831.
- Krum SA, Miranda GA, Lin C, Lane TF. BRCA1 Associates with Processive RNA Polymerase II. J Biol Chem. 2003;278:52012–52020.
- Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, Liu M, Lonigro R, Prensner JR, Tomlins SA, et al. PARP-1 Inhibition as a Targeted Strategy to Treat Ewing's Sarcoma. Cancer Research. 2012;72:1608–1613.
- Vormoor B, Curtin, N. J. Poly(ADP-ribose) polymerase inhibitors in Ewing sarcoma. Curr Opin Oncol. 2014;26:428–433.
- Paronetto MP, Miñana B, Valcárcel J. The Ewing Sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell. 2011;43:353–368.
- Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–124.
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts S, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218.