
ABSTRACT
For cancer cells to survive during extracellular matrix (ECM)-detachment, they must inhibit anoikis and rectify metabolic deficiencies that lead to the induction of non-apoptotic cell death. Here, we highlight and discuss our recent study implicating receptor-interacting protein kinase-1 (RIPK1) in the induction of mitophagy, the production of reactive oxygen species (ROS) and the consequent elimination of ECM-detached cells.
A vast majority of epithelial cell types are dependent on integrin-mediated attachment to extracellular matrix (ECM) to receive growth-potentiating signals that support biosynthesis, proliferation, and survival. Interestingly, lack of ECM-attachment results in the induction of anoikis, an apoptotic, caspase-dependent cell death program. In contrast, cancer cells often inhibit anoikis in a fashion that enables their survival and ultimately facilitates tumor progression and metastasis.Citation1 Anoikis inhibition is not sufficient for long term survival of ECM-detached cancer cells as additional, anoikis-independent cellular changes can profoundly impact survival. More specifically, the survival of ECM-detached cells requires recalibration of redox metabolism in a fashion that ameliorates lethal reactive oxygen species (ROS) and abrogates consequent non-apoptotic cell death.Citation2,Citation3 While a number of studies have unveiled strategies utilized by cancer cells to evade anoikis,Citation1,Citation4,Citation5 the precise molecular mechanisms that contribute to and execute non-apoptotic death during ECM-detachment remain poorly understood.
Over the last several years, it has become apparent that necrosis is not solely induced by acute cellular stress or injury.Citation6 Rather, necrosis can be activated through a genetic program as a result of the activation of kinases in the receptor-interacting protein kinase (RIPK) family. Intriguingly, several studies have linked ROS to RIPK-dependent programmed necrosis. Thus, our recent study () sought to investigate the possibility that activation of RIPK family members contributes to non-apoptotic death during ECM-detachment.Citation7 In support of this supposition, we discovered a sizable increase in RIPK1 levels and activation as a result of ECM-detachment. Furthermore, inhibition of RIPK1 activity with necrostatin-1 (Nec1) in MCF-10A cells led to enhanced cell viability during ECM-detachment and improved luminal filling in 3-dimensional cultures of mammary acini. We next extended this analysis into cancer cell lines, and discovered that treatment of multiple, distinct cancer cell lines with Nec1 led to significantly improved viability during ECM-detachment and enhanced anchorage-independent growth in soft agar. Given these findings, we reasoned that RIPK1 may compromise the viability of ECM-detached cells through the activation of RIPK3 and mixed lineage kinase domain like pseudokinase (MLKL) (known effectors of RIPK1-mediated necrosis).Citation6 Unexpectedly, we found that inhibiting RIPK3 or MLKL (either pharmacologically or via shRNA) failed to impact the viability of ECM-detached cells. This surprising result led us to conclude that RIPK3 and MLKL are dispensable for RIPK1-mediated cell death during ECM-detachment.
Figure 1. RIPK1 mediated induction of mitophagy during ECM detachment. When a cell detaches from extracellular matrix (ECM) (here depicted as the red cell detached from the blue ECM), Cylindromatosis (CYLD) will deubiquitinate receptor interacting protein kinase 1 (RIPK1) to promote its stability. Subsequently, RIPK1 will become fully active via autophosphorylation at S161. Next, RIPK1 will bind to and interact with phosphoglycerate mutase family member 5 (PGAM5) and PTEN-induced putative kinase 1 (PINK1). The formation of this complex will protect PINK1 from Presenilin associated rhomboid like (PARL) protease-mediated degradation, enabling PINK1 to promote mitophagy. As a consequence of mitophagy induction, ECM-detached cells experience diminished isocitrate dehydrogenase 2 (IDH2)-mediated NADPH production in the mitochondria and the subsequent elevation in mitochondrial ROS levels leads to non-apoptotic cell death.
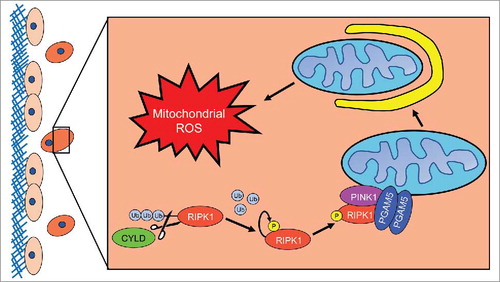
Thus, we focused our studies on alternate downstream effectors of RIPK1 signaling that may impact viability. Previous work suggested that phosphoglycerate mutase family member 5 (PGAM5), a mitochondrial phosphatase, could function downstream of RIPK-mediated signaling in a fashion that impacts mitochondrial integrity and cell viability.Citation8 Interestingly, shRNA-mediated reduction in PGAM5 led to a significant elevation in the viability of ECM-detached cells and to robust luminal filling in 3-dimensional cultures of mammary acini. Given the localization of PGAM5 at the mitochondria and the fact that recent studies have implicated PGAM5 in the regulation of PTEN-induced putative kinase 1 (PINK1)-mediated mitophagy,Citation9 we assessed the impact of RIPK1 inhibition on PINK1. We found that RIPK1 inhibition led to Presenilin associated rhomboid like (PARL) protease-mediated proteolytic cleavage of PINK1 and consequent loss of its enzymatic activity. Given these data and the observed importance of PGAM5, we reasoned that the formation of a multi-protein complex containing RIPK1, PGAM5, and PINK1 could interfere with PARL-mediated PINK1 cleavage. In support of this possibility, we found that Nec1 treatment blocks the interactions of these proteins and renders PINK1 susceptible to PARL-mediated cleavage.
To more directly assess the possibility that RIPK1/PGAM5-mediated stabilization of PINK1 results in mitophagy during ECM-detachment, we evaluated the expression of multiple mitochondrial markers. Indeed, these markers are substantially downregulated during ECM-detachment and rescued by inhibition of RIPK1. Similarly, quantification of mitochondrial number using MitoTracker revealed a detachment-mediated loss of mitochondrial number that was rescued by Nec1 treatment. Given the aforementioned studies suggesting that elevated levels of ROS could trigger cell death during ECM-detachment, we speculated that mitophagy induction could contribute to ROS production. Indeed, our studies revealed that a significant elevation in mitochondrial ROS as a consequence of mitophagy induction.
These findings led us to conduct additional studies aimed at better understanding the mechanism by which mitophagy leads to mitochondrial ROS production. Recently, an unconventional pathway used by ECM-detached cells to adapt to ROS was uncovered.Citation10 This pathway involves reductive formation of citrate by isocitrate dehydrogenase 1 (IDH1), consequent NADPH production in mitochondria by isocitrate dehydrogenase 2 (IDH2), and resulting mitigation of mitochondrial ROS. We thus hypothesized that a consequence of RIPK1/PGAM5-mediated mitophagy is diminished capacity for mitochondrial IDH2 to counteract ROS. Indeed, the enhanced viability resulting from RIPK1 inhibition in ECM-detached cells requires the enzymatic activity of mitochondrial IDH2.
Furthermore, our data raise the possibility that RIPK1/PGAM5-mediated cell death in ECM-detached cells could function to antagonize the survival of cancer cells in vivo. Interestingly, we discovered that H460 cells deficient in RIPK1 or PGAM5 could more efficiently form tumors in the lungs of immunocompromised mice following intravenous injection. We also examined publically available lung cancer patient microarray data sets and discovered that low RIPK1 expression in lung cancer patients correlates with decreased overall and progression-free survival. In aggregate, these findings suggest that RIPK1/PGAM5-mediated mitophagy may function in a tumor suppressive capacity to eliminate invasive, metastatic ECM-detached cancer cells.
These findings uncover an unexpected role for RIPK1 in compromising the viability of ECM-detached cells and suggest that RIPK1-mediated mitophagy functions to suppress tumor formation. Future studies aimed at delineating which cell types are most susceptible to RIPK1-mediated cell death are paramount. Equally important is to better understand the mechanism by which mitophagy induction causes ROS production and cell death in cancer cells. It is well appreciated that mitophagy can function to remove defective mitochondria, but our data suggest that, in some cancers, there may be a selection for cancer cells with loss of function mutations in mitophagy. In aggregate, additional studies that build upon our recent findings are critical to understanding how best to leverage this potential vulnerability in a therapeutic context.
Abbreviations
| ECM | = | extracellular matrix |
| Nec1 | = | necrostatin-1 |
| RIPK1 | = | receptor-interacting protein kinase-1 |
| ROS | = | reactive oxygen species |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14:632–41. doi:10.1038/nrc3789. PMID:25098270.
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–13. doi:10.1038/nature08268. PMID:19693011.
- Mason JA, Hagel KR, Hawk MA, Schafer ZT. Metabolism during ECM Detachment: Achilles Heel of Cancer Cells? Trends Cancer. 2017;3:475–81. doi:10.1016/j.trecan.2017.04.009. PMID:28718402.
- Mason JA, Davison-Versagli CA, Leliaert AK, Pape DJ, McCallister C, Zuo J, Durbin SM, Buchheit CL, Zhang S, Schafer ZT. Oncogenic Ras differentially regulates metabolism and anoikis in extracellular matrix-detached cells. Cell Death Differ. 2016;23:1271–82. doi:10.1038/cdd.2016.15. PMID:26915296.
- Weigel KJ, Jakimenko A, Conti BA, Chapman SE, Kaliney WJ, Leevy WM, Champion MM, Schafer ZT. CAF-secreted IGFBPs regulate breast cancer cell anoikis. Mol Cancer Res. 2014;12:855–66. doi:10.1158/1541-7786.MCR-14-0090. PMID:24803643.
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47. doi:10.1038/nrm3737. PMID:24452471.
- Hawk MA, Gorsuch CL, Fagan P, Lee C, Kim SE, Hamann JC, Mason JA, Weigel KJ, Tsegaye MA, Shen L, et al. RIPK1-mediated induction of mitophagy compromises the viability of extracellular-matrix-detached cells. Nat Cell Biol. 2018;20:272–84. doi:10.1038/s41556-018-0034-2. PMID:29459781.
- Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–43. doi:10.1016/j.cell.2011.11.030. PMID:22265414.
- Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, Zhang Y, Dawson VL, Dawson TM, Lenardo M. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson's-like movement disorder. Nat Commun. 2014;5:4930. doi:10.1038/ncomms5930. PMID:25222142.
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532:255–8. doi:10.1038/nature17393. PMID:27049945.