
ABSTRACT
Pharmaceutical inhibition of the Human Epidermal growth factor Receptor 2 (HER2) oncogene has dramatically improved outcomes in HER2-amplified breast cancers. However, monotherapy HER2 inhibitors are not effective. We have recently reported that a co-amplified microRNA within the HER2 amplicon, leads to activation of the oncogene Myeloid Cell Leukemia-1 (MCL-1), tempering cell death responses to HER2 inhibitors. Importantly, HER2 inhibitors are sensitized to cell death by the addition of pharmacological MCL-1 inhibitors, which are entering clinical trials.
The Human Epidermal growth factor Receptor 2 (HER2) oncogene is located on chromosome 17 at position 17q12. Focal amplification of 17q12 is found in 20% of breast cancer cases. Overexpression of the HER2 receptor (encoded by the HER2 gene) results in its homo- or hetero-dimerization, its autophosphorylation and the consequent propagation of an oncogenic signaling primarily through the PI3K/AKT/mTOR and the MAPK/ERK pathways. Over the past several years, different targeted molecules have been designed that selectively target specific Receptor Tyrosine Kinases (RTKs), and in cancers addicted to these RTKs, these therapies induce apoptosis through the modulation of the B-Cell Leukemia/Lymphoma 2 (BCL-2) family members. In fact, cancers that fail to undergo a robust apoptotic response are the cancers that have poorer overall responses, shorter progression free survivals, and poorer overall survivals. In one of these paradigms, Epidermal Growth Factor Receptor (EGFR)-mutant lung cancer, low expression level of the BH3-only protein, B-cell lymphoma 2 Interacting Mediator of cell death (BIM), results in a lack of apoptotic responses and therefore reflects poor clinical outcomes.Citation1-Citation3 Furthermore, we have recently shown that the well-known resistance of mesenchymal-like EGFR-mutant lung cancers to EGFR inhibitors is at least partly due to the downregulation of BIM by the mesenchymal transcriptional repressor, Zinc Finger E-Box Binding Homeobox 1 (ZEB1).Citation4 When there is sufficient BIM, like in many epithelial-like EGFR-mutant lung cancers, EGFR inhibitors lead to robust responses.Citation1,Citation2 Interestingly though, in HER2-amplified breast cancers, HER2 inhibitors although widely useful as adjuvant therapy, are ineffective as monotherapy, despite the ability to suppress oncogenic signaling in these cancers.
These data suggested to us that a uniform apoptotic block may be present in HER2-amplified cancers. By evaluating BCL-2 family RNA levels in several breast cancer tumor datasets, including The Cancer Genome Atlas (TCGA), we found that in fact this was the case: excessively low levels of the gene encoding Phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, also known as NOXA), a BH3-only protein, were apparent.
Interestingly, HER2 manipulation did not affect NOXA expression: After using specific siRNA sequences against HER2 in HER2-amplified breast cancer cell lines, or by ectopically overexpressing HER2 cDNA in HER2-negative breast cancer cell lines (driven by a viral promoter), we concluded that there is no causative link between HER2 receptor signaling and NOXA downregulation. This suggested that the inverse correlation between HER2 and NOXA was not a result of HER2 regulation of NOXA. The first clue to the actual connection between the two was when we observed that NOXA levels were the lowest in HER2+/estrogen receptor (ER)- breast cancers, implying a role of the ER in the expression of NOXA.
We identified an intronic microRNA, termed miR-4728,Citation5 that is hosted in the HER2 gene. Since its expression depends on the same promoter of the HER2 transcript, it gets co-amplified with HER2. Importantly, a target of miR-4728 is the mRNA of Estrogen Receptor α (ESR1).Citation6 We found that manipulation of miR-4728 levels was sufficient to inversely regulate ERα in breast cancer cell lines. Importantly, loss or gain of ERα expression resulted in correlative loss or gain of expression of its downstream target, NOXA ().
Figure 1. The primary transcript of miR-4728 is co-amplified together with its host gene (HER2) in the nucleus and pre-miR-4728 is transported into the cytoplasm for further processing to become mature miR-4728 that inhibits ESR1 mRNA and consequently leads to the ERα-mediated downregulation of NOXA.
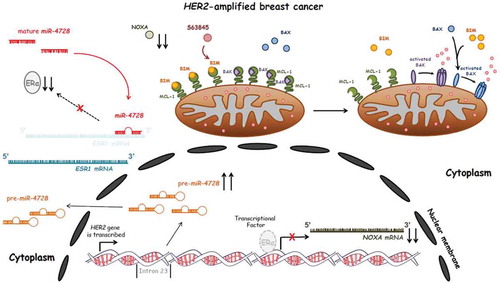
NOXA serves as an antagonist of the bona fide oncogene MCL-1, an important and emerging drug target. For example, we reported elevated NOXA in MYCN-amplified neuroblastomas,Citation7 which enhanced the susceptibility of these tumors to BCL-2/BCL-XL inhibitors via NOXA-mediated MCL-1 neutralization. Indeed, the deficiency of NOXA can be sufficient to prolong the accumulation of fellow pro-apoptotic BCL-2 members like BIM on the outer mitochondrial membrane, thus altering the threshold of apoptosis and conferring resistance to targeted therapies such as HER2 inhibitors.
Importantly, MCL-1 specific inhibitors have now been developed, and are entering clinical trials (e.g. NCT0267545). To test whether MCL-1 inhibition was sufficient to sensitize to HER2 inhibitors, we treated HER2-amplified breast cancers with the combination of the two drugs, using two different MCL-1 inhibitors (A1210477, AbbVie, and S63845, Servier). We noted disruption of MCL-1 – BIM and MCL-1 – Bcl-2 homologous antagonist/killer (BAK) complexes in the cells co-treated with the MCL-1 inhibitor, and noted combination killing that was primarily dependent on both BIM and BAK (). Impressively, in two mouse models of HER2-amplified breast cancer, lapatinib and S63845 combined to have potent anti-tumor activity. These data are further support of this combination, which efficacy was first reported in HER2-amplified models using the HER2 inhibitor trastuzumab in combination with S63845Citation8; in addition, the authors reported that killing of cells with HER2i/S63845 was at least partially dependent on BIM and BAK, which was consistent with our observations as well.
While there have been numerous cases reported in the literature in which a microRNA is implicated in cancer progression, either by enhancing or by blocking tumorigenesis, to our knowledge, this is the first time that a specific co-amplified microRNA (or any protein-coding gene) causes intrinsic resistance to a targeted therapy directed against the oncogenic signaling driven by the amplification of its host gene. It is noteworthy that other targeted therapies in other oncogenic paradigms—like inhibitors directed against EGFR in EGFR-mutant lung cancer or BRAF in BRAF-mutant melanoma – do not share the same concerns of co-amplified genes, as these genes are active by mutation.
To summarize, the phenomenon of intronic microRNAs to support the function of their host genes has already been described.Citation9 However, in our study,Citation10 we have found that a microRNA species which is co-amplified within the HER2 amplicon, and in this case the HER2 gene itself, mitigates the efficacy of HER2 inhibitors. With many other actionable cancer drivers activated by amplification, such as EGFR in Head and Neck cancers and colorectal cancers, a careful assessment of the other co-amplified genes may provide important mechanistic insights (and possibly biomarkers) for intrinsic resistance to the therapies directed against these cancer drivers. This is particularly relevant given the extreme heterogeneity of responses to EGFR in these two cancers. In addition, as direct inhibitors of Kirsten Rat Sarcoma (KRAS) and myelocytomatosis (MYC) family members are actively being sought, we should be mindful that, similarly, other co-amplified genes may contribute to the response or resistance to these inhibitors.
Acknowledgements
This work was supported by NCI grant K22CA175276 to ACF and a Mary Kay Foundation grant (ACF).
References
- Faber AC, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, Incio J, Digumarthy SR, Pollack SF, Song Y, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1( 4):352–65. doi:10.1158/2159-8290.CD-11-0106. PMID:22145099
- Karachaliou N, Codony-Servat J, Teixidó C, Pilotto S, Drozdowskyj A, Codony-Servat C, Giménez-Capitán A, Molina-Vila MA, Bertrán-Alamillo J, Gervais R, et al. BIM and mTOR expression levels predict outcome to erlotinib in EGFR-mutant non-small-cell lung cancer. Sci Rep. 2015;5: 17499. doi:10.1038/srep17499. PMID:26639561
- Cardona AF, Rojas L, Wills B, Arrieta O, Carranza H, Vargas C, Otero J, Corrales-Rodriguez L, Martín C, Reguart N, et al. BIM deletion polymorphisms in Hispanic patients with non-small cell lung cancer carriers of EGFR mutations. Oncotarget. 2016;7( 42):68933–42. doi:10.18632/oncotarget.12112. PMID:27926478
- Song KA, Niederst MJ, Lochmann TL, Hata AN, Kitai H, Ham J, Floros KV, Hicks MA, Hu H, Mulvey HE, et al. Epithelial-to-Mesenchymal Transition Antagonizes Response to Targeted Therapies in Lung Cancer by Suppressing BIM. Clin Cancer Res. 2018;24( 1):197–208. doi:10.1158/1078-0432.CCR-17-1577. PMID:29051323
- Persson H, Kvist A, Rego N, Staaf J, Vallon-Christersson J, Luts L, Loman N, Jonsson G, Naya H, Hoglund M, et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer Res. 2011;71( 1):78–86. doi:10.1158/0008-5472.CAN-10-1869. PMID:21199797
- Newie I, Søkilde R, Persson H, Grabau D, Rego N, Kvist A, von Stedingk K, Axelson H, Borg Å, Vallon-Christersson J, et al. The HER2-encoded miR-4728-3p regulates ESR1 through a non-canonical internal seed interaction. PloS one. 2014;9 (5):e97200. doi:10.1371/journal.pone.0097200. PMID:24828673
- Ham J, Costa C2, Sano R3, Lochmann TL4, Sennott EM, Patel NU, Dastur A, Gomez-Caraballo M, Krytska K, Hata AN, Floros KV, et al. Exploitation of the Apoptosis-Primed State of MYCN-Amplified Neuroblastoma to Develop a Potent and Specific Targeted Therapy Combination. Cancer cell. 2016;29( 2):159–72. doi:10.1016/j.ccell.2016.01.002. PMID:26859456
- Merino D, Whittle JR, Vaillant F, Serrano A, Gong JN, Giner G, Maragno AL, Chanrion M, Schneider E, Pal B, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med. 2017;9(401):eaam7049. doi:10.1126/scitranslmed.aam7049. PMID:28768804
- Lutter D, Marr C, Krumsiek J, Lang EW, Theis FJ. Intronic microRNAs support their host genes by mediating synergistic and antagonistic regulatory effects. BMC Genomics. 2010; 11:224. doi:10.1186/1471-2164-11-224. PMID:20370903
- Floros KV, Lochmann TL1, Hu B2, Monterrubio C3, 4, Hughes MT, Wells JD, Morales CB, Ghotra MS, Costa C, Souers AJ, Boikos SA, et al. Coamplification of miR-4728 protects HER2-amplified breast cancers from targeted therapy. Proc Natl Acad Sci U S A. 2018;115(11):E2594-E2603. PMID:29476008