
ABSTRACT
Isoform selection of pyruvate kinase M (PKM), a glycolytic enzyme, influences fates of glucose-derived carbons in cellular metabolic networks. We recently developed novel mouse lines to study PKM isoform function and identified PKM1 as a potential target in a subset of human lung cancers. This work provides new insight into cancer metabolism.
The glycolytic enzyme pyruvate kinase M (PKM) exists as two isoforms: PKM1, which is constitutively active and promotes glucose catabolism, and PKM2, which is activated only in response to increased levels of allosteric activator(s). The latter property ensures that PKM2 will maintain a lower rate of glycolysis flux to limit glucose oxidation relative to PKM1. Generally, expression of PKM1 and PKM2 is mutually exclusive in a given cell type, as isoforms are generated by alternative splicing of transcripts from a single gene. It has long been assumed that PKM2 is expressed embryonically in most organs/tissues and then replaced by PKM1 as development proceeds, although evidence for this switch is inconclusive. It has also been assumed that PKM1 expression is not compatible with proliferation and that PKM2 is the only isoform expressed in dividing cells. However, mouse genetic analysis has revealed discrepant findings: PKM2-specific knockout (PKM2-KO) mice, created by deletion of a PKM2-specific exon, develop normally and exhibit enhanced tumorigenesis in several experimental models.Citation1,Citation2 Importantly, PKM2-KO mice also display compensatory and partial expression of the more active isoform PKM1, at varying levels.Citation1-Citation3 A major unanswered question then became, whether oncogenic phenotypes seen in PKM2-KO mice were due to increased or decreased PK activity? More specifically, it was unclear whether PKM2 activity is cancer-promoting or -suppressing. We addressed this question by evaluating PKM isoform function in novel mouse models.Citation4
Specifically, we used a knock-in (KI) rather than knock-out approach and developed mutant mouse lines specifically expressing PKM1 (PKM1-KI) or PKM2 (PKM2-KI) from the endogenous Pkm locus, a strategy that allowed tissue/cell type-specific PKM expression. Both types of homozygous KI mice developed normally, indicating that loss of one PKM isoform has minimal effect if the other isoform is present in sufficient levels. Interestingly, working in both genetically- and chemically-induced models, we observed significantly enhanced tumor formation in PKM1-KI animals compared to WT or PKM2-KI mice. PKM1-KI mice showed enhanced KRAS-induced lung tumorigenesis and 7,12-dimethylbenz[a]anthracene-initiated various tumors, including liver tumors. Accordingly, others have reported enhanced liver tumorigenesis in PKM2-KO mice, an outcome attributable to a non-cell autonomous mechanism since these tumor cells express neither PKM1 nor PKM2.Citation1 In contrast, we observed PKM1-positive hepatocellular carcinoma nodules and several other types of tumors expressing PKM1 in PKM1-KI mice. Strikingly, PKM1-expressing tumor cells grew more rapidly than did PKM2-KI or WT cells in transplantation models. Based on this analysis, we conclude that PKM1 rather than PKM2 promotes tumor growth in a cell-intrinsic manner.
We next asked how PKM1 alters cellular metabolism. Using 13C-glucose tracer experiments we showed that PKM1-expressing cells exhibit higher glucose flux into the lactate and the tricarboxylic acid cycle. Of note is that PKM1 did not impede biosynthetic glucose metabolism through the pentose phosphate pathway or nucleotide synthesis. Another key point was that PKM1-KI cells showed lower levels of total lactate, whereas PKM1 increased levels of glucose-derived lactate relative to WT and PKM2-KI, similar to a previous observation by Christofk.Citation5 Further study is needed to resolve this enigma, although impaired anaplerotic use of glutamine by PKM1-KI cells could underlie these outcomes.
When mitochondrial function is impaired, active aerobic metabolism can produce unfavorable by-products, such as reactive oxygen species (ROS). Thus, we analyzed mitochondrial properties in Pkm-KI contexts. Surprisingly, PKM2-KI cells contained more mitochondria than did PKM1-KI cells, but mitochondria were functionally impaired as revealed by decreased membrane potential and higher ROS production. This finding suggests that unhealthy mitochondria accumulate in PKM2 cells. Given that autophagy eliminates damaged mitochondria (in a process called mitophagyCitation6), we speculated that PKM1 activates autophagy/mitophagy more efficiently than does PKM2, contributing to malignancy. In support of this hypothesis, PKM1 cells showed higher autophagic activity than did PKM2 cells, and ablation of autophagy-related 7 (Atg7) gene, encoding an essential factor for autophagy, largely decreased growth of PKM1-expressing tumor cells. Overall, we conclude that autophagy/mitophagy is more active in PKM1-KI than in PKM2-KI cells, potentially conferring a metabolic advantage to PKM1 cells. Future studies should address mechanisms by which PKM1 activates autophagy.
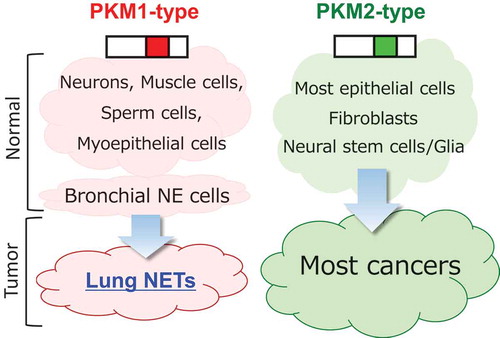
Although we found that PKM1 can play a tumor-enhancing role if expressed in cancer cells, most human cancer cells express PKM2. Thus, we monitored cells of various cells of origin to determine whether tumors arising from PKM1-positive cells express PKM1 (). Pulmonary neuroendocrine tumors (NETs) include a spectrum of tumors from low-grade carcinoid to high-grade large-cell neuroendocrine carcinoma (LCNEC) and small-cell lung cancer (SCLC). NETs account for about 15% of lung cancers, and patient prognosis in this subset is relatively poor compared to other lung cancers. Importantly, NETs reportedly originate from NE cells, which are PKM1-positive.Citation7 Strikingly, we observed that pulmonary NETs express Pkm1 and that Pkm1 expression is required for SCLC cell proliferation.
Figure 1. Tumor cells of origin and pyruvate kinase M (PKM) isoforms.
Both lung neuroendocrine tumors (NETs) and their cells of origin (bronchial neuroendocrine (NE) cells) show high PKM1 expression, whereas other most tumor cells and their cells/tissues of origin express PKM2.
In summary, we report that expression of PKM1, rather than PKM2, activates glucose metabolism and boosts tumor growth cell-intrinsically in various models. Our results challenge the idea that limiting glucose catabolism by PKM2 is required to sustain biosynthetic metabolism and cell proliferation. However, we do not exclude the possibility that PKM2 confers some advantage(s) to tumor cells non-cell autonomously, as in case of autophagy, which plays a complex, sometimes dual role in carcinogenesis.Citation8 Nevertheless, PKM1 is potential new target for lung NET, one of the deadliest cancers known.Citation9,Citation10
Disclosure statement
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks T, et al. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev. 2016a;30:1020–1033. doi:10.1101/gad.278549.116.
- Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409. doi:10.1016/j.cell.2013.09.025.
- Lunt SY, Muralidhar V, Hosios AM, Israelsen WJ, Gui DY, Newhouse L, Ogrodzinski M, Hecht V, Xu K, Acevedo PN, et al. Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol Cell. 2015;57:95–107. doi:10.1016/j.molcel.2014.10.027.
- Morita M, Sato T, Nomura M, Sakamoto Y, Inoue Y, Tanaka R, Ito S, Kurosawa K, Yamaguchi K, Sugiura Y, et al. Pkm1 confers metabolic advantages and promotes cell-autonomous tumor cell growth. Cancer Cell. 2018;33:355–367. doi:10.1016/j.ccell.2018.02.004.
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi:10.1038/nature06734.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi:10.1016/j.cell.2011.10.026.
- Asselin-Labat ML, Filby CE. Adult lung stem cells and their contribution to lung tumourigenesis. Open Biol. 2012;2:120094. doi:10.1098/rsob.120094.
- Pietrocola F, Manuel Bravo-San PJ, Galluzzi L, Kroemer G. Autophagy in natural and therapy-drivenanticancer immunosurveillance. Autophagy. 2017;13:2163–2170. doi:10.1080/15548627.2017.1310356.
- Pietanza MC, Byers LA, Minna JD, Rudin CM. Small cell lung cancer: will recent progress lead to improved outcomes? Clin Cancer Res. 2015;21:2244–2255. doi:10.1158/1078-0432.CCR-14-2958.
- Naidoo J, Santos-Zabala ML, Iyriboz T, Woo KM, Sima CS, Fiore JJ, Kris MG, Riely GJ, Lito P, Iqbal A, et al. Large Cell Neuroendocrine Carcinoma of the Lung: clinico-Pathologic Features, Treatment, and Outcomes. Clin Lung Cancer. 2016;17:e121–129. doi:10.1016/j.cllc.2016.01.003.