
ABSTRACT
Loss-of-function mutations of the chromatin regulator ATRX (α-thalassemia mental retardation X-linked) occur frequently in diffuse gliomas, but the molecular mechanisms by which ATRX inactivation promotes oncogenesis remain unclear. We recently reported that Atrx deficiency drives glioma-relevant phenotypes, such as increased motility and astrocytic differentiation profiles, by directly modulating epigenomic lanscapes in glioma cells of origin. Our work has significant implications on the role of epigenetic regulator dysfunction in the oncogenic process.
Diffuse gliomas represent the most common adult and pediatric brain tumors. While they are histologically and molecularly heterogeneous, they are all incurable at present, due to both their wide infiltration into surrounding normal brain, and their tendency to relapse in the face of intensive treatment with surgery, radiation, and chemotherapyCitation1. Recent comprehensive analyses integrated histopathologic, molecular and prognostic features of diffuse gliomas establishing important correlations between somatic driver alterations, molecular disease classification, and clinical outcomeCitation2. These advances have laid the groundwork for the development of more effective treatment strategies, efforts that will require an improved understanding of the unique molecular features driving the pathogenesis of individual glioma subclasses.
Inactivating mutations in the chromatin remodeling gene ATRX (α-thalassemia mental retardation X-linked) represent defining molecular alterations in major subgroups of both adult and pediatric glioma that tend to exhibit morphologic and immunohistochemical features of astrocytes and are thus classified as “astrocytomas”. In these tumors, ATRX deficiency invariably co-occurs with mutations in tumor protein p53 (TP53, best known as p53), and in genes encoding either isocitrate dehydrogenase enzymes (IDH1 and IDH2) in adults or H3.3 histone monomers (H3F3A and HIST13HB) in childrenCitation3–Citation5. So far, ATRX inactivation in cancer has been solely correlated with a telomerase-independent mechanism of telomere maintenance known as alternative lengthening of telomeres (ALT)Citation6. However, the downstream effects of ATRX deficiency on cellular epigenomic landscapes and their pathogenic consequences are almost entirely unknown.
For the first time, we recently reported that Atrx deficiency influences the expression of specific gene sets that drive glioma-relevant phenotypes by directly modulating epigenomic profiles in glioma cells of originCitation7. We modeled the cellular and molecular context of ATRX-mutant gliomagenesis by inactivating Atrx in p53-intact and deficient murine neuroepithelial progenitors (mNPCs), and observed that Atrx deficiency, particularly when combined with p53 loss, promotes in mNPCs cell migration while also shifting the expression of differentiation markers toward an astrocytic lineage. These phenotypes recapitulate two defining features of infiltrating astrocytomas and their acquisition was accompanied by large shifts in transcriptional profiles that strongly correlated with known gene expression signatures derived from ATRX-mutant gliomasCitation3,Citation4. These findings indicate that transcriptional alterations induced by Atrx deficiency in mNPCs and their downstream functional sequelae are highly reminiscent of those occurring in ATRX-mutant gliomas.
We went on to characterize the molecular basis of Atrx-deficient phenotypes in mNPCs. In particular, we demonstrated that the increased cellular motility arising with Atrx deficiency is, at least in part, due to upregulation of G protein subunit alpha 13 (Gna13), an upstream effector of ras homolog family member A (RhoA) GTPase signalingCitation8. Moreover, we found that Atrx deficiency disrupted the expression of crucial astrocytic makers and master regulators such as, glial fibrillary acidic protein (Gfap), inhibitor of DNA binding 3 (Id3) and signal transducer and activator of transcription 3 (Stat3). Validating these mechanistic findings in ATRX-mutant human gliomas and primary patient-derived glioma stem cell lines, provided further support for the disease relevance of our discoveries.
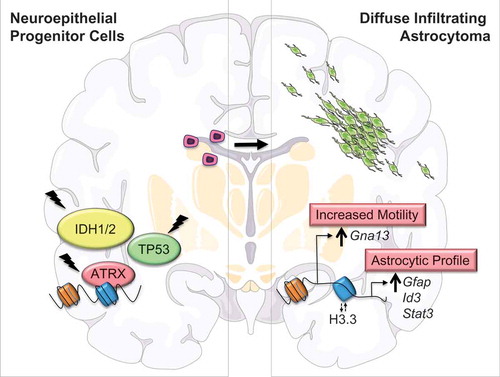
Integrating the transcriptional changes described above with genome-wide Atrx distribution and chromatin accessibility profiles occurring with Atrx deficiency demonstrated that Atrx loss directly impacts gene expression through global epigenomic remodeling. Particularly significant correlations were observed for key genes driving disease-defining phenotypes, such as Gfap and Gna13, whose promoter regions exhibited Atrx binding sites as well as shifts in chromatin accessibility following Atrx inactivation. Moreover, Atrx deficiency at these loci was associated with disruptions in H3.3 histone content, consistent with an established mechanism by which ATRX regulates chromatin structure and organization. Taken together, these findings indicate that Atrx loss modulates chromatin composition primarily in the immediate vicinity of vacant Atrx binding sites, dysregulating local gene expression and promoting phenotypic behavior typical of diffuse astrocytic gliomas ().
Figure 1. Epigenomic and transcriptional dysregulation occurring with ATRX deficiency drive disease-defining phenotypes in glioma cells of origin. ATRX (α-thalassemia mental retardation X-linked) loss of function mutations, together with IDH1/2 (isocitrate dehydrogenase enzymes 1 and 2) and TP53 (tumor protein p53) mutations, are defining molecular alterations characterizing the diffusely infiltrating astrocytomas. We demonstrated that Atrx inactivation alters chromatin structure and accessibility in the immediate vicinity of vacant Atrx binding sites (blue), in part due to shifts in the incorporation of the H3.3 histone variant. These changes induce the misexpression of locally situated genes, promoting the acquisition of disease-defining cellular phenotypes, such as motility and induction of astrocytic gene expression profiles.
The significance of our work lies in its characterization of novel mechanisms by which mutational disruptions involving epigenetic regulator networks can directly mediate cancerous cellular behavior. In doing so, we also describe targetable molecular pathways mediating key phenotypes in a malignant, incurable disease. Finally, we provide concrete evidence that the gliomagenic effects of ATRX deficiency are not limited to genomic instability and ALT, which have received the lion’s share of attention from the cancer research community to date, but also include broad epigenomic dysfunction, consistent with the established role of ATRX as a regulator of chromatin state and compositionCitation9.
In recent years, it has become increasingly evident that dysregulated epigenetic processes can play central roles in cancer onset and progression, diffuse glioma includedCitation1. The reversibility of epigenetic modifications renders them suitable for pharmacological interventions. As such, they are now considered attractive therapeutic targets. Inhibitors of chromatin modulating enzymes, like the histone methyltransferases DOT1 like histone lysine methyltransferase (DOT1L) and enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) as well as the demethylase lysine demethylase 1A (KDM1A), have already reached early-stage clinical trials for cancer therapyCitation10. We are confident that similarly addressing the epigenetic effects of ATRX deficiency has the potential to transform personalized therapy for malignant gliomas, particularly those harboring ATRX mutations.
Disclosure statement
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Huse JT, Aldape KD. The molecular landscape of diffuse glioma and prospects for biomarker development. Expert Opin Med Diagn. 2013;7(6):573–587. doi:10.1517/17530059.2013.846321.
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016; 164(3): 550–563.doi:10.1016/j.cell.2015.12.028.
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, et al. Comprehensive, integrative genomic analysis of diffuse lower- grade gliomas. N Engl J Med. 2015;372(26): 2481–2498. doi:10.1056/NEJMoa1402121.
- Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT. Whole exome sequencing identified ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012;3(10):1194–1203. doi:10.18632/oncotarget.689.
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482(7384): 226–231.doi:10.1038/nature10833.
- Heaphy CM, De Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011; 333(6041): 425.doi:10.1126/science.1207313.
- Danussi C, Bose P, Parthasarathy PT, Silberman PC, Van Arnam JS, Vitucci M, Tang OY, Heguy A, Wang Y, Chan TA, et al. Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat Commun. 2018; 9(1): 1057.doi:10.1038/s41467-018-03476-6.
- Kelly P, Casey PJ, Meigs TE. Biologic functions of the G12 subfamily of heterotrimeric g proteins: growth, migration, and metastasis. Biochemistry. 2007;46(23):6677–6687. doi:10.1021/bi700235f.
- Watson LA, Goldberg H, Bérubé NG. Emerging roles of ATRX in cancer. Epigenomics. 2015;7(8):1365–1378. doi:10.2217/epi.15.82.
- Morera L, Lübbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics. 2016;8:.57. doi:10.1186/s13148-016-0223-4.