
ABSTRACT
The dual phosphatase CDC25 has recently been identified as a target for diverse triple-negative breast cancers including RB1/PTEN/P53-deficient tumors. Moreover, CDC25 inhibitors effectively synergize with PI3K inhibitors to suppress tumor growth. We discuss these findings and the challenges that lie ahead in bringing CDC25 inhibitors to the clinic.
Authors’ view
Somatic alterations in genes that control cell cycle progression represent a major hallmark of cancer.Citation1,Citation2 Loss of cell cycle control is driven by inactivation of the tumor suppressor RB1 either by direct mutations/deletions in the gene or by phosphorylation and inactivation of the protein, pRb, by Cyclin Dependent Kinase (CDK) 2, 4 and 6. In breast cancer, pRb is inactivated by hyper-phosphorylation in luminal and HER2+ subtypes, whereas the RB1 gene is frequently disrupted together with p53 (TP53) in triple-negative breast cancer (TNBC). The tumor suppressor PTEN is also frequently inactivated together with p53, and in some cases with both RB1 and p53 in TNBC. TNBC is highly aggressive and no targeted therapy is currently available.
Hyper-phosphorylation of pRb is amenable to therapeutic intervention using CDK4/6 inhibitors such as Palbociclib (PD-0332991), which show promising results in clinical trials. However, RB1-deficiency confers resistance to anti-CDK4/6 therapy.Citation3 Thus, RB1 gene loss poses a major therapeutic challenge. This realization and the fact that RB1-loss is not directly druggable have prompted a worldwide search for alternative mechanisms to target RB1-deficiency.
One approach involves the analysis of new pathways downstream of RB1, which are amenable to therapeutic interventions. Indeed, two groups have recently shown that RB1-loss couples the induction of cell division with mitochondrial protein translation (MPT) and oxidative phosphorylation (OXPHOS). This in turn leads to increased sensitivity to antagonists of MPT and OXPHOS as well as to inhibitors of interleukin 6.Citation4,Citation5
In another approach, discussed herein, RB1-deficient TNBC lines were screened for drugs that can effectively suppress cell growth. In one study, drug screens of isogenic human breast cancer lines in which RB1 gene was disrupted by CRISPR-mediated technology, identified CHK1 and PLK1 inhibitors as synthetically lethal with RB1-deficiency.Citation6 In our study, drug screens of human TNBC lines with combined mutations in RB1, PTEN and p53, or of primary tumor cells isolated from mouse models of mammary-specific Rb plus p53 or Pten plus p53 conditional knockout, identified the CDC25 phosphatase as a common target.Citation7 Importantly, inhibition of CDC25 effectively killed RB1/P53-deficient TNBC cells that are refractory to CDK4/6 plus CDK2 inhibitors. Moreover, RNAi-mediated knockdown of CDC25A plus CDC25B mimicked the effect of CDC25 inhibitors and obliterated growth of TNBC cells.
CDC25 regulates transitions through the G1/S and G2/M phases of the cell cycle.Citation8 For example, it promotes entry into mitosis by dephosphorylating tyrosine 15 (Y15) and activating CDK1 downstream of ATR and CHK1 (). The WEE1 kinase antagonizes CDC25 by phosphorylating the same Y15 in CDK1, thereby blocking progression into mitosis. DNA damage induces ATR upstream of CHK1, which in turn phosphorylates and activates WEE1 and inhibits CDC25 (). Thus, ATR/CHK1/WEE1 inhibitors promote passage through the DNA damage/CDK1 checkpoint, whereas CDC25 (and CDK1) antagonists have the opposite effect. ATR/CHK1/WEE1 inhibitors are thought to suppress tumor growth by an “over the cliff” mechanism through which premature or forced entry into M phase induces mitotic catastrophe. But how does inhibition of CDC25 lead to cell death? In normal cells, CDC25 inhibition prevents CDK1 activation leading to cell cycle arrest (). However, in addition to its regulation of CDK1, CDC25 also suppresses several apoptotic pathways, which are unleashed in response to its inhibition (). For example, CDC25 dephosphorylates Apoptosis Signal-regulating Kinase (ASK1) on Thr838. CDC25 blockade induces pThr838-ASK1, which phosphorylates and activates several pro-apoptotic substrates including Stress-activated protein kinases (SAPK)/Jun amino-terminal kinases (JNK). Interestingly, we found that CDC25 antagonists induced phosphorylation of ASK1 in TNBC (MS and EZ, unpublished) as well as phosphorylation of Ser73-cJUN, a downstream target of JNK.Citation7 CDC25 likely blocks other pro-apoptotic pathways either directly (like ASK1) or indirectly. As an example for the latter, CDC25 induces CDK1/cyclin B1, which phosphorylates caspase 9 on an inhibitory Thr125. Thus, inhibition of CDK1 via CDC25 antagonists activates this key caspase leading to cell demise. It would be important to identify additional pro-apoptotic targets of CDC25, as well as the environmental contexts, e.g. hypoxia and anchorage-independent growth, in which CDC25 maintains survival of TNBC.
Figure 1. Molecular rationale for CDC25-targeting therapies. (A) In proliferating cells, CDC25 dephosphorylates Y15-CDK1 allowing for G2 to M progression. (B) In response to DNA damage, cells arrest at the G1/S (not shown) and/or G2/M transitions through activation of ATM/ATR, which induce CHK1/2, leading to phosphorylation of several targets. Phosphorylation of WEE1 stimulates this kinase to phosphorylate tyrosine (Y) 15 on CDK1 leading to cell cycle arrest. Phosphorylation of the CDC25 phosphatase inhibits its dephosphorylation of Y15-CDK1, further blocking G2/M progression. (C) CDC25 inhibitors unleash apoptotic pathways directly downstream of CDC25 (e.g. ASK1) or downstream of CDK1 (e.g. pCASPASE 9), leading to cell demise in diverse triple-negative breast cancers (TNBCs). (D) Combined treatment with CHK1 plus WEE1 inhibitors is synergistic through a mechanism yet to be established. (E) Sustained inhibition of CDC25 induces pSer473-PKB/AKT, likely as a feedback loop or epigenetic adaptation downstream of PI3K signaling that prolongs cell survival. (F) Combined treatment with CDC25 plus PI3K inhibitors is highly synergistic, leading to effective killing of TNBC.
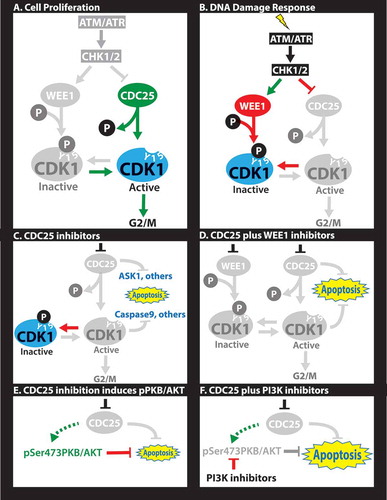
Combination treatments with CDC25 plus WEE1 inhibitors showed excellent synergy in suppressing growth of TNBC cells (). This was somewhat unexpected as these proteins antagonize each other by exerting opposite effects on Y15-CDK1. We also observed weak synergy at low concentrations between CDC25 and CDK1 inhibitors, but antagonistic effects at higher concentrations. This is in accordance with the notion that CDC25 acts upstream not only of CDK1 but also of other factors like ASK1. Thus, CDC25-based therapy offers a unique avenue to combat TNBC. Furthermore, as CDC25 inhibition suppresses growth of normal cells, it may be advantageous over ATM/ATR, CHK1/2 or WEE1 inhibitors that may drive aberrant proliferation and clonal expansion of pre-neoplastic cells.
CDC25 was previously considered as a therapeutic target for diverse types of malignancies including breast cancer, but interest in this target has waned for two main reasons. First, the type of patients that would benefit from anti-CDC25 therapy has never been defined. Our observation that TNBCs are highly sensitive to CDC25 inhibitors should renew interest in CDC25-based therapy. Such therapy may also be tested for other cancers with similarity to TNBC and/or with loss of RB1 plus p53 such as serous cervical carcinoma, small-cell lung carcinoma, pancreatic adenocarcinomas, and perhaps retinoblastoma. Second, there are currently no available CDC25 inhibitors with clinical utility. Of the existing inhibitors, the most potent are quinonoid derivatives, which irreversibly oxidize the cysteine residue in the catalytic domain of CDC25 and likely hit other substrates. They all have IC50s in the micromolar range compared to nanomolar range of inhibitors for other targets (kinase) on this pathway (e.g. CHK1/2, WEE1). A recent computational screening of over 2 million compounds by docking into the CDC25B active site identified a plethora of CDC25 inhibitors, some of which are reversible, but they also have IC50s in the micromolar range and their activity in vivo has not been determined.Citation9 An alternative direction would be to develop allosteric inhibitors for CDC25, as was recently accomplished for the SHP2 phosphatase. However, this approach is challenged by variability in the N-terminus of CDC25A and CDC25B due to differential splicing, the need to target both phosphatases, and lack of crystal structure for the N-terminus of either protein. Further computational analysis with improved algorithm for drug design, high throughput screens of hundreds of thousands of new agents, and thorough analysis by in vitro and xenograft assays may uncover CDC25 inhibitors with improved clinical utility.
A complementary approach for CDC25-based therapy would involve combination therapy using moderate concentrations of existing CDC25 inhibitors. Effective combination therapy may involve targeting other vulnerabilities in each specific tumor as part of precision medicine regimens, or on blocking escape routes of tumor cells in response to anti-CDC25 therapy. Such escape routes may entail pre-existing or de novo mutations that bypass CDC25 inhibition, or epigenetic adaptation to anti-CDC25 therapy. In this regard, long-term (24 hrs) inhibition of CDC25 resulted in strong induction of pSer473AKT/PKB, a survival signal downstream of PI3K (Ref.Citation7,. In accordance, combinations of CDC25 plus PI3K inhibitors were highly synergistic both in vitro and in xenotransplantation model of TNBC (. Elucidating the mechanism by which pSer473AKT/PKB is induced following CDC25 inhibition may uncover new therapeutic approaches to preempt its activation. Moreover, non-biased sensitization screens may identify new potent combination therapy(ies) with available anti-CDC25 antagonists. Finally, combinations with new therapies for TNBCs such as epigenetic (e.g. JQ1), eEF2K inhibitors or disulfiramCitation10 may also prove effective.
In conclusion, CDC25 emerges as a central survival factor in TNBC, whose inhibition alone or in combination with other drugs such as WEE1 or PI3K inhibitors induces apoptosis in TNBC, independent of the status of RB1, PTEN or p53. Understanding the mechanism by which CDC25 inhibits cell death, the generation of potent new CDC25 inhibitors, and the development of effective combination therapy with CDC25 inhibitors are some of the challenges that lie ahead in bringing CDC25-based therapy to the clinic.
Disclosure of potential conflicts of interest
No potential conflicts of interest are disclosed.
Additional information
Funding
References
- Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, Wang S, Herschkowitz JI, Egan SE, Perou CM, et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle. 2011;10(10):1563–1570. doi:10.4161/cc.10.10.15703.
- Zacksenhaus E, Shrestha M, Liu JC, Vorobieva I, Chung PED, Ju Y, Nir U, Jiang Z. Mitochondrial OXPHOS induced by RB1 deficiency in breast cancer: implications for anabolic metabolism, stemness, and metastasis. Trends Cancer. 2017;3(11):768–779. doi:10.1016/j.trecan.2017.09.002.
- Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, Dubois J, Nagy RJ, Lanman RB, Iafrate AJ, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. 2018; 29(3):640–645. doi:10.1093/annonc/mdx784.
- Jones RA, Robinson TJ, Liu JC, Shrestha M, Voisin V, Ju Y, Chung PE, Pellecchia G, Fell VL, Bae S, et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J Clin Invest. 2016; 126(10):3739–3757. doi:10.1172/JCI81568.
- Kitajima S, Yoshida A, Kohno S, Li F, Suzuki S, Nagatani N, Nishimoto Y, Sasaki N, Muranaka H, Wan Y, et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene. 2017; 36(36):5145–5157. doi:10.1038/onc.2017.124.
- Witkiewicz AK, Chung S, Brough R, Vail P, Franco J, Lord CJ, Knudsen ES. Targeting the vulnerability of RB tumor suppressor loss in triple-negative breast cancer. Cell Rep. 2018;22(5):1185–1199. doi:10.1016/j.celrep.2018.01.022.
- Liu JC, Granieri L, Shrestha M, Wang DY, Vorobieva I, Rubie EA, Jones R, Ju Y, Pellecchia G, Jiang Z, et al. Identification of CDC25 as a common therapeutic target for triple-negative breast cancer. Cell Rep. 2018; 23(1):112–126. doi:10.1016/j.celrep.2018.03.039.
- Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7(7):495–507. doi:10.1038/nrc2169.
- Lavecchia A, Di Giovanni C, Pesapane A, Montuori N, Ragno P, Martucci NM, Masullo M, De Vendittis E, Novellino E. Discovery of new inhibitors of Cdc25B dual specificity phosphatases by structure-based virtual screening. J Med Chem. 2012;55(9):4142–4158. doi:10.1021/jm201624h.
- Robinson TJ, Pai M, Liu JC, Vizeacoumar F, Sun T, Egan SE, Datti A, Huang J, Zacksenhaus E. High-throughput screen identifies disulfiram as a potential therapeutic for triple-negative breast cancer cells: interaction with IQ motif-containing factors. Cell Cycle. 2013;12(18):3013–3024. doi:10.4161/cc.26063.