
ABSTRACT
Genetic lineage tracing in cell type-specific mouse models of T-cell acute lymphoblastic leukemia (T-ALL) have revealed that tumor cell identity is imposed by expression of the oncogene Lim Domain Only 2 (LMO2), rather than by the target cell phenotype. This approach allowed to identify that secondary genomic alterations, like Notch1 mutations, appeared late and only took place within the thymus during T-ALL development. These concepts are therefore critical for the development of modern therapies aimed at curing T-ALL.
T-cell acute lymphoblastic leukemia (T-ALL) accounts for about 15 to 25% of acute lymphoblastic leukemias in children and adults. T-ALL remains a significant clinical problem given the inability to cure many patients, and the significant toxicity of current therapies, highlighting the need for better treatment strategies. Molecular and genomic studies have created a renewed interest in the NOTCH signaling pathway as a therapeutic target for the treatment of T-ALL (). In spite of these advances, we still do not understand the mechanisms leading to the origin of T-ALL cells sufficiently well to have an impact on T-ALL mortality.
Figure 1. Rationale for choosing therapeutic targets for T-ALL treatment.
Acquired NOTCH1 activating mutations are frequently present in T-ALL, so it seems to be an ideal therapeutic target to treat the sickness. But the recent deeply understanding of the generation and evolution of the disease is moving the focus for selecting therapeutic targets. We have recently shown that Lmo2 is able to primed hematopoietic stem/progenitor cells (HS/PCs) for T-cell malignancy and is not necessary anymore once the tumor-T cell identity is established. Secondary mutations like NOTCH1 are late events in leukemia evolution. In this sense, targeting NOTCH1 may not have the desirable therapeutic complete respond and we would need to add Notch1-independent targets for therapy. T-ALL: T-cell acute lymphoblastic leukemia.
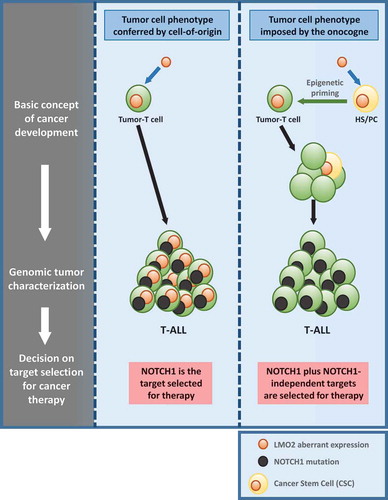
During leukemogenesis a normal cell acquires a new but inappropriate (malignant) identity to give rise to a clonal aberrant population. This is only possible if the oncogenic event initiating cancer had an inherent reprogramming capacity, to be able to lead to the change in cellular identity.Citation1 LMO2 is one of the most frequent drivers of childhood T-ALLCitation2,Citation3 and has been recently identified as one of the six transcription factors required for reprogramming committed murine blood cells into induced hematopoietic stem cells.Citation4 This observation led us to hypothesize that LMO2 may function as a ‘hit-and-run’ oncogene that acts at an early stage of T-cell development to reprogram hematopoietic stem/progenitor cells (HS/PCs) for T-cell malignancy. To test this hypothesis, we generated genetic lineage tracing strains of mice with restricted expression of this oncogene within HS/PCs.Citation5 Despite lack of Lmo2 protein expression within T-cells, these mice develop aggressive and clonal T-ALL. The resulting T-ALL lacked Lmo2 and its targeted-gene expression, and were histologically, transcriptionally and genetically similar to human LMO2 driven T-ALL in which LMO2 alterations are frequently observed.Citation5 These results therefore confirm that activity of the Lmo2 oncogene restricted to HS/PCs can induce malignancies in mice that are of a T-cell stage of differentiation. However, the permissiveness for development of T-ALL seems to be associated with wider windows of differentiation than previously appreciated. Restricted Cre-mediated activation of Lmo2 at different stages of B-cell development induced systematically and unexpectedly T-ALL that closely resembled those of their natural counterparts.Citation5 Biological barriers exist to prevent cells from changing their identity in this manner in order to avoid the risk of malignant transformation.Citation1,Citation6,Citation7 In B-cell malignancies has been shown that loss of p53 is a frequent occurrence and facilitates the pathological reprogramming to a malignant B-cell phenotype.Citation8,Citation9 Similarly, a significant proportion of T-ALL in all our murine models carried p53 loss-of-function mutations facilitating pathological reprogramming to a malignant T-cell phenotype.Citation5 In conclusion, we have provided the first evidence of a ‘hit-and-run’ role for Lmo2 in the oncogenesis of T-ALL. The implication of these findings led us to propose that T-ALL is the result of an inappropriate lineage-decision making process occurring via a reprogramming-like mechanism. Thus, oncogenes that initiate tumor formation seem to be dispensable for tumor cell survival and/or tumor progression.
As to the capacity of the LMO2 to reprogram cells, it is generally accepted that tumoral progression is a multi-hit process.Citation1 In this case different aspects of normal cellular biology are progressively altered to finally give rise to a full-blown tumor. Thus we next trace where and when these second hits appeared and found that the secondary mutations, like Notch1 mutations, appeared late and only took place within the thymus during T-ALL development.Citation5 This may provide an explanation for the failure of some modern targeted therapies to clear tumor stem cells, despite being effective agents against evolved tumor cells. For instance, NOTCH1 has emerged as a potentially important therapeutic target in recent years given its frequent involvement in T-ALL and the fact that specific NOTCH1 inhibitors can kill T-ALL cells in vitro and in vivo. The fact that NOTCH1 mutations are not present within the T-ALL precursor cells will limit the therapeutic efficacy of targeting NOTCH1 (). If this prediction is correct and given the possibility to carry out single-cell sequencing, then it should be possible to probe that NOTCH1 mutations are late mutations in human T-ALL. Using targeted single-cell sequencing has been provided recent evidence that NOTCH1 mutations, as predicted, were typically late events.Citation10 From the therapeutic standpoint this would indicate the need to combine NOTCH1 inhibitors with drugs that can kill these NOTCH1-independent tumor cells (). These concepts are therefore central to the full understanding of T-ALL genesis and critical for the development of precise and modern therapies aimed at directly curing T-ALL.
Acknowledgments
We are indebted to all members of our groups for useful discussions and for their critical reading of the manuscript. J.H. has been supported by the German Cancer Aid (Project 110997 and Translational Oncology Program 70112951), the German Carreras Foundation(DJCLS 02R/2016), the Kinderkrebsstiftung (2016/17) and the “Elterninitiative Kinderkrebsklinik e.V. Düsseldorf”. AB has been supported by the German Children’s Cancer Foundation and the Federal Ministry of Education and Research, Bonn, Germany. Research in ISG group is partially supported by FEDER and by MINECO (SAF2012-32810, SAF2015-64420-R and Red de Excelencia Consolider OncoBIO SAF2014-57791-REDC), by Junta de Castilla y León (BIO/SA51/15, CSI001U14, UIC-017, and CSI001U16). ISG lab is a member of the EuroSyStem and the DECIDE Network funded by the European Union under the FP7 program. AB and ISG have been supported by the German Carreras Foundation (DJCLS R13/26). GRH was supported by FSE-Conserjería de Educación de la Junta de Castilla y León (CSI001-15). Research in CVD group is partially supported by FEDER, “Miguel Servet” Grant (CP14/00082 - AES 2013-2016) from the Instituto de Salud Carlos III (Ministerio de Economía y Competitividad), “Fondo de Investigaciones Sanitarias/Instituto de Salud Carlos III” (PI17/00167) and by the Lady Tata International Award for Research in Leukaemia 2016-2017.
Additional information
Funding
References
- Vicente-Duenas C, Hauer J, Cobaleda C, Borkhardt A, Sanchez-Garcia I. 2018. Epigenetic Priming in Cancer Initiation. Trends Cancer. 4:408–417. PMID:29860985. doi:10.1016/j.trecan.2018.04.007.
- Van Vlierberghe P, van Grotel M, Beverloo HB, Lee C, Helgason T, Buijs-Gladdines J, Passier M, van Wering ER, Veerman AJ, Kamps WA, et al. The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood. 2006;108:3520–3529. PMID:16873670. doi:10.1182/blood-2006-04-019927.
- Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, McCastlain K, Edmonson M, Pounds SB, Shi L, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–1218. PMID:28671688. doi:10.1038/ng.3909.
- Riddell J, Gazit R, Garrison BS, Guo G, Saadatpour A, Mandal PK, Ebina W, Volchkov P, Yuan GC, Orkin SH, et al. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–564. PMID:24766805. doi:10.1016/j.cell.2014.04.006.
- Garcia-Ramirez I, Bhatia S, Rodriguez-Hernandez G, Gonzalez-Herrero I, Walter C, Gonzalez De Tena-Davila S, Parvin S, Haas O, Woessmann W, Stanulla M, et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018; PMID:29880602. doi:10.15252/embj.201798783.
- Rodriguez-Hernandez G, Hauer J, Martin-Lorenzo A, Schafer D, Bartenhagen C, Garcia-Ramirez I, Auer F, Gonzalez-Herrero I, Ruiz-Roca L, Gombert M, et al. Infection Exposure Promotes ETV6-RUNX1 Precursor B-cell Leukemia via Impaired H3K4 Demethylases. Cancer Res. 2017;77:4365–4377. PMID:28630052. doi:10.1158/0008-5472.CAN-17-0701.
- Martin-Lorenzo A, Auer F, Chan LN, Garcia-Ramirez I, Gonzalez-Herrero I, Rodriguez-Hernandez G, Bartenhagen C, Dugas M, Gombert M, Ginzel S, et al. Loss of Pax5 Exploits Sca1-BCR-ABL(p190) Susceptibility to Confer the Metabolic Shift Essential for pB-ALL. Cancer Res. 2018;78:2669–2679. PMID:29490943. doi:10.1158/0008-5472.CAN-17-3262.
- Vicente-Duenas C, Fontan L, Gonzalez-Herrero I, Romero-Camarero I, Segura V, Aznar MA, Alonso-Escudero E, Campos-Sanchez E, Ruiz-Roca L, Barajas-Diego M, et al. Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc Natl Acad Sci U S A. 2012;109:10534–10539. PMID:22689981. doi:10.1073/pnas.1204127109.
- Vicente-Duenas C, Romero-Camarero I, Gonzalez-Herrero I, Alonso-Escudero E, Abollo-Jimenez F, Jiang X, Gutierrez NC, Orfao A, Marin N, Villar LM, et al. A novel molecular mechanism involved in multiple myeloma development revealed by targeting MafB to haematopoietic progenitors. Embo J. 2012;31:3704–3717. PMID:22903061. doi:10.1038/emboj.2012.227.
- De Bie J, Demeyer S, Alberti-Servera L, Geerdens E, Segers H, Broux M, De Keersmaecker K, Michaux L, Vandenberghe P, Voet T, et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia. 2018; PMID:29740158. doi:10.1038/s41375-018-0127-8.