
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The fast and continuous growth of multidisciplinary approaches, consisting of the combination of various experimental techniques and computational methods tuned on purpose to depict and predict material properties and performance, is motivated by the need to design new superior devices for effective applications in a great variety of industrial sectors. This strategy not only produces excellent results but is also fundamental to drive the research towards innovative and cost-effective production systems. In this short review we outline the major steps of our theoretical modelling activity connected to the experimental gas-phase synthesis of silicon nanoparticles (SiNPs) in plasmas reactors, limiting the discussion to the long-scale simulations based on the classical reactive description (ReaxFF methodology) for reproducing the whole process, perturbations included. The huge computational workload at the quantum chemistry level, required to prepare the training data, calibrate the parameters and validate the procedure, is not reported for the sake of conciseness. The excellent agreement of the simulations results with the experimental data and the sound explanations of the observed phenomena suggest that the ReaxFF-based paradigm is a reliable approach capable of studying these types of materials on time and size scales corresponding to realistic scenarios.
Major steps of the theoretical modelling activity connected to the experimental gas-phase synthesis of silicon nanoparticles (SiNPs) in plasmas reactors.
Graphical abstract
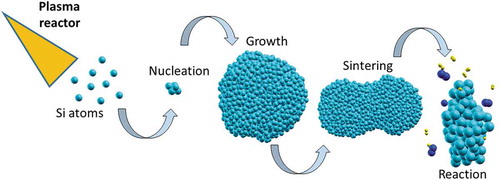
1. Introduction
The capability of modeling structural and dynamical behaviour of different types of materials at the atomic level, on time and size scales close to experiments, is extremely useful for designing novel devices with improved performance at reduced production costs. This is because punctual characterizations in selected portions of the chosen components can be essential to gain a deeper understanding of the effects of specific modifications on the whole materials properties and to suggest new engineering strategies to modulate their response. As a matter of fact, model size and simulation time bias the choice of the modelling approaches, which, in the case of materials, are usually those based on pre-parametrized force fields and mainly consist of molecular dynamics simulations, Monte Carlo methods and combinations of the two. Several studies and accurate investigations have demonstrated that classical force fields modelled on pairwise potentials to simulate metals and semiconductors behaviours are often unsuitable for reproducing a number of properties, such as the ratio between the cohesive energy and the melting temperature, the ratio between the vacancy formation energy and the cohesive energy, the ratio between the elastic constants C12/C44, and surface characteristics [Citation1]. It is also recognized that when dealing with various types of materials, reactivity is very often a key element for the correct operation of the assembled components. This is why the capability to simulate bond breaking and formation was incorporated in the force fields expressions by developing empirical reactive descriptions in assorted varieties (the Brenner-Tersoff, Stillinger–Weber, AIREBO and ReaxFF potentials are just a few). These reactive force fields, which contain connection dependent terms, were prepared to reproduce realistically the materials performance and were progressively upgraded, over the years, by the addition of new features and training structures. Indeed, to prepare appropriate parameter sets, the force fields should be trained against sample molecular configurations obtained and evaluated through first principle calculations, and/or experimental data. Then, particular selections, such as the training structures, their ‘weights’ (or importance of each configuration in the parameter optimization) and the type of contributions being reproduced by the fitting algorithm (geometry, energy difference, etc.) should be used to tune the specificity of the force field and its transferability.
One of the most powerful reactive interatomic potentials used nowadays for treating reliably a great variety of materials is the ReaxFF approach that was developed by van Duin and co-workers and published in 2001 in a paper focused on hydrocarbons [Citation2]. Since then the ReaxFF development tree (see in Ref[Citation3].) has grown impressively and branched out into an elevated ‘multicolored’ crown. In the case of silicon, the first ReaxFF description appeared in 2003 [Citation4] and was essentially used to predict morphology, properties, interfacial behaviour and hydrolysis of materials made of silicon and silicon oxides. In a second step, the focus was shifted to crack propagation in a silicon single crystal and the ReaxFF approach was included in a hybrid multiscale scheme where it was combined with the Tersoff potential. This synergistic combination had an improved performance in terms of computational speed and accuracy and was capable of reproducing fractures (by means of the ReaxFF component) and elastic deformations (by means of the Tersoff representation) of silicon accurately [Citation5,Citation6]. Other studies explored the behaviour of silicon-based compounds such as silicon carbide [Citation7,Citation8], silanes [Citation9], silicates [Citation10,Citation11], whereas more recent investigations were focused on the functionalization of silicon surfaces with organic molecules [Citation12,Citation13]. It follows that silicon parameters can be found in many different ReaxFF force fields, also because silicon is contained in various types of composites [Citation14]. However, in each case, its parameters have been revised and readapted to reproduce accurately the target material and the related reactions/interactions.
Over the last few years our research activity has been addressed to the computational description of the silicon nanoparticles (SiNPs) growth mechanism in homogeneous and heterogeneous plasma environments. Calculations were carried out by means of quantum chemistry methods and molecular dynamics simulations based on specific parametrizations of a ReaxFF force field suitable to describe high temperature conditions found in plasma reactors. This research was conducted in close collaboration with experimental scientists interested in optimizing the production processes [Citation15] and was aimed at disclosing the atomic details of the nanoparticles formation and self-interactions starting from embryo-subparticles, which in a monoatomic gas act as nucleation centres of the homogeneous nucleation mechanisms. The simulations, starting from the already decomposed chemical precursors, were capable of reproducing the evolution of the NPs structures and their dynamics, also when the condensation proceeded in high-temperature chemically reacting gas flows (usually involving hydrogen, nitrogen or even oxygen). As evidenced in the literature, a complex plasma consists of various elements such as ions, neutral atoms, molecules, small particles and is nurtured by the collisions between the different charged or activated chemical components [Citation16]. During the re-condensation phase ultrafine particles are produced. The operation conditions include very often nitrogen and hydrogen as carrier gases but also as cooling (N2) or reducing (H2) agents. This happens in Si nanopowders production as well. SiNPs are one of the most important components of the semiconducting devices for microelectronic applications [Citation17,Citation18] and thus they require large-scale fabrication in various morphologies with tuned and efficient techniques. The effects due to the gases used in the fabrication could be fundamental to regulate feeding and production rates. Thus, they were specifically investigated through a series of simulations focused on the adsorption of nitrogen and hydrogen on preformed silicon clusters. This is because it was useful to single out the maximum effects due to each of these molecular gases on the silicon clusters when the other plasma species were not present. These were just the first stages of the design of a more complex scenario. In this review we outline all the stages of this long study by illustrating concisely the force field optimization protocols developed in our group and the series of molecular dynamics simulations in non-reactive and reactive environments, where reactivity refers to the presence of other species, other than Si, that can play a role in the NPs formation.
2. ReaxFF parameters and parametrization strategies
To generate a ReaxFF force field capable of reproducing surface effects, which are very important for clusters and NPs, we included in the training set the lowest minimum energy configurations of relatively small Si clusters, obtained through quantum chemistry (QC) optimizations at the DFT level (Quantum ESPRESSO program [Citation19]). The smallest and the biggest clusters contained 2 and 36 atoms, respectively, and all the sizes in between, were examined in detail proceeding in two-atom steps. More specifically, the selection of the representative structures for each family was based on geometry optimization and Basin-Hopping (BH) [Citation20,Citation21], which was repeated five times at most (approximately 500 steps). At each step, the structure of the cluster was randomly changed, optimized and then accepted or rejected according to the Metropolis algorithm (kT between 0.5 and 1.0 eV). All the details regarding the search procedures and the selection of the minima to be included in the training set can be found in [Citation22,Citation23]. Once the training set was arranged a number of force field parameters that should be optimized were identified (24 in all), a cost function for comparison with the QC values was defined, by choosing geometric and energetic terms, and then specific weights were assigned to all the included conformers. Beside the original sequential algorithm for parameters optimization, where one parameter was optimized at a time [Citation24], available in the stand-alone version of the ReaxFF program, two global optimization algorithms were tested, namely the Monte Carlo Force Field optimizer MCFF [Citation25] contained in the ADF suite of programs [Citation26] and the Genetic Algorithm (GA) implemented in the GA based Reactive Force Field (GARFfield) program [Citation8]. Although these more advanced methods are almost completely automated and less biased while they minimize the error function [Citation27], they require large High Performance computational resources for parallel optimizations and a number of starting choices defining ranges and convergence. As far as the computer power was concerned it was found that the MCFF algorithm was less demanding (in terms of error evaluations) than GA, could handle satisfactorily the numerical noise in the error function and escape higher local minima to some extent. Furthermore, the comparison of the methods behaviour in relation to the Si training set revealed that both the sequential and MCFF optimizations produced combinations of parameters of similar performance in terms of structural predictions and transferability when validated on larger clusters, whereas the GA implementation generated parameters that resulted in smaller deviations of the training data but could not confidently be used to describe structures outside the training set [Citation23]. The final force field, optimized for carrying out simulations of SiNPs, was the one obtained by means of the serial optimization procedure and was further expanded to include the effects of the carrier/cooling/reducing gasses. This was obtained by adding all the parameters describing hydrogen, nitrogen and their interactions with Si to the force field.
3. Nucleation and growth: the formation of SiNPs in an argon atmosphere
To simulate the formation of SiNPs in an argon plasma at the experimental reactor conditions, classical molecular dynamics (MD) simulations based on the optimal force field selected after the first parametrization stage (pure silicon), were performed by means of the LAMMPS code [Citation28]. All the simulations were carried out in the NVT ensemble at different temperature (Berendsen’s thermostat) and composition. Before starting the production simulations, preliminary studies were conducted to calibrate required parameters such as dimension of the simulation box, thermostat, type of ensemble, integration time step, parametrization of the force field, temperature of the reactor, gas pressure (atomic density in the simulation box) and composition of the gas mixture, which could affect SiNPs growth rates and aggregation modes (for a detailed discussion see refs [Citation23,Citation29].).
Indeed, within a reactor, nucleation of SiNPs takes place in a sovrasaturation region where the temperature of the hot plasma of Si atoms abruptly drops; the pressure of the gas phase is higher than that one at the thermodynamic equilibrium and the gas condenses into small liquid droplets (embryo SiNPs). The minimum dimension of these embryo clusters resulted from the condensation can be derived from the classical nucleation theory [Citation30] (in the hypothesis of gas-liquid equilibrium) and corresponds to the balancing between stabilizing cohesive (bulk) and de-stabilizing surface energies. This classical view of the nucleation process is based on the hypothesis that the gas and the liquid states are always in equilibrium. However, this disagrees with the atomistic view where instabilities of the system induce the formation of embryo SiNPs at any temperature if two or more atoms favourably collide and form chemical bonds. The concept of a critical dimension, which states that the cluster is unstable, becomes false as in the whole range of temperatures, characteristic of plasma reactors, stable silicon dimers are identified. In this new perspective, the attention is shifted from nucleation (which starts from a dimer) to growth, where velocity and modality are strongly connected to the thermodynamic conditions of the reactor (composition of the gas mixture and temperature).
Given these premises, to quantify the growth rate, we have introduced the concept of inception time for a cluster of size N as the minimum time at which this cluster appears in the simulation box during the dynamics. Essentially, the inception time can be seen as the average time needed by the system to form a cluster of size N. From a computational point of view, the inception time has strong statistical fluctuations and can be estimated only by averaging the results of several independent simulations. For a given size N, it is quite intuitive that inception time increases with the decrease of the system atomic density (hence the pressure) and temperature. The former relation is due to the fact that a decrease in the Si density (realized either through a reduction of the absolute pressure or a dilution of Si in Ar) determines a decrease in the encounter probability between Si atoms and, as a consequence, an increase in inception time.
To prove these two assumptions, we considered inception time trends and growth mechanisms of two different systems where the composition of the gas mixture was 50% Si – 50% Ar (C1) and 25% Si – 75% Ar (C2) in a temperature range between 1800 and 2400 K.
C1 inception times for N = 5, 10, 15, 20, 25, 30, 35 and 40 as a function of temperature are shown in . From the plot, a monotone rise of the inception time for any size and temperature is apparent. Each point of the plot of ) has been obtained by averaging over three independent MD simulations. We can thus make the following assumption:
Figure 1. (a) inception times (ns) for N = 5 (purple), 10 (green), 15 (cyan), 20 (orange), 25 (yellow), 30 (blue), 35 (red) and 40 (black) as a function of the temperature for a system containing 50% Si and 50% Ar (C1). Inception time for a cluster of size N is defined as the minimum time at which this cluster appears in the simulation box during the dynamics. (b) function aP(N) (in arbitrary units, for a definition of this function, refer to Equation (1)–(2) for the two different compositions: C1 (purple dots) and C2 (blue triangles).
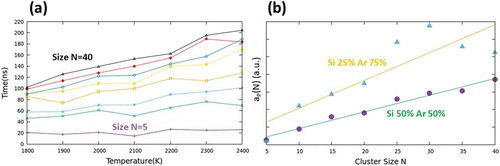
where t(N,T) is the time needed to generate a cluster of size N at a temperature T and aP(N) and bP(N) are two functions which depend on N and parametrically on pressure P (hence composition/density of the system). In order to gain some information of a possible analytic form of the aP(N) function, we have plotted the angular coefficients of the curves of ) as a function of N, see purple dots in ). From the latter plot, it is quite reasonable to express a linear relation between aP(N) and N:
where αP and βP are two functions which parametrically depend on P.
The analogous plots corresponding to C2 are not shown in ) and only the linear regression corresponding to the averaged values is displayed () – cyan triangles and yellow line). The comparison between C2 and C1 demonstrates that a density decrease in the number of reactive species leads to an increase in inception times in the whole range, meaning that αP is a function which increases when decreasing P. The linear fitting, however, seems not appropriate when the cluster size is larger than 20. This is because the fluctuations that affect the aggregates in a more diluted system are larger and require a more extended sample for obtaining reliable averages.
To give a more complete description of the simulation scenario of the SiNPs growth, the attention was focused on the single steps of the process. It was found that inside the plasma, particle growth could take place either through the addition of single atoms to a cluster or through the aggregation of small clusters with various sizes. These modes can coexist and could be both detected by tracking the evolution of the number of atoms forming the selected particle ()).
Figure 2. (a) evolution of the number of atoms during the simulation for two characteristic clusters of finite size 86 Si atoms (green line) and 115 Si atoms (red line); the simulation has been carried out at P = 1.00 atm and T = 2000 K in a mixture made of 25% Si and 75% Ar. (b) Sintering plot for two particles made of about 1000 atoms (radius about 1.7 nm) on a temperature range between 1000 and 1500 K; initial and final structures (cyan balls).
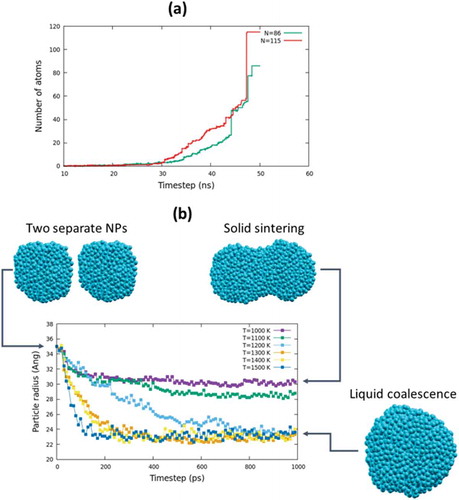
Inspection of the evolution of the number of atoms of two different particles revealed a similar growth trend made of two different stages atom-by-atom addition (indicated by the smooth profile of the growing curve) and then (after 45 ns, in this specific case) cluster aggregation. This is a clear indication of the coalescence of pre-formed particles and poses another issue related to the sintering/coalescence of the clusters that come close to one another. As a matter of fact, a prominent role during this process is played by the physical state of the particles [Citation31–Citation33]. In fact, when working above the melting temperature of bulk silicon (which according to experiments is approximately 1700 K), all the SiNPs are liquid droplets that quickly rearrange after addition of individual atoms or larger pre-formed aggregates. This is the regime of liquid coalescence, where the typical time to complete sintering is well below 1 ns for particles up to 3–4 nm (estimated radius). A completely different scenario emerges instead when the NPs are solid, and typical times to complete sintering could be order of magnitudes larger than those of the liquid coalescence [Citation31]. To verify this assumption, the sintering plots of two approaching SiNPs (about 1.7 nm in radius, i.e. made by about 1000 Si atoms) investigated in a temperature range between 1000 and 1500 K is shown in ). As it is well known [Citation34], the melting points of finite NPs are lower than the melting point of the bulk, although an exact relation between particle size and melting point is not straightforward. In the present investigation, the sintering process has been monitored through the evolution of the radius of an imaginary sphere containing the two approaching particles during the sintering/coalescence [Citation33]. According to the plot, shown in ), the quick evolution of the radius (from about 3.5 nm to about 2.3 nm), which takes place at temperature above 1300 K, is a clear indication that in this temperature range the NPs are liquid and coalescence is taking place. Lowering the temperature, coalescence is remarkably cooled down at T = 1200 K, (in around 1 ns); for lower temperatures, a solid sintering regime is observed as the radius, after an initial decrease due to the formation of a meniscus between the two SiNPs (see the picture showing the structure), does not change, indicating the on-set of very slow motions of solid re-arrangements. This analysis indicates the importance of the operando temperature in the formation of NPs, as the physical state of the growing particles influence their final morphology and indirectly establishes the melting point of NPs as a function of their size. According to ReaxFF predictions, the melting point of a SiNP of radius about 1.7 nm is located between 1100 and 1300 K.
4. Nucleation and growth simulations: formation of SiNPs in a mixed Ar/H2/N2 environment
The early stages of the gas phase condensation of SiNPs in a low-temperature plasma, where H2 and N2 molecules are present, were simulated to qualitatively and quantitatively estimate how these gas species influence the nucleation and growth processes. The selected conditions also agree with the parameters chosen for microwave plasma production of metal nano powders [Citation35], where the final products derive from the chemical reactions taking place in the gas flow (nitrogen in this case) under microwave irradiation. According to the literature this type of synthesis is relatively stable, can be carried out at ambient pressure and at a temperature in the 580–1200 K range. In microwave plasma reactors, it is during the recondensation in the gas phase that the chemical reactions with the plasma species take place and determine the characteristics of the final product. Then, the product is cooled, collected and filtered. Thus, the residence time in the plasma and the specific processing parameters determine the mean particle size.
The computational model built for reproducing such a recondensation procedure contains Si atoms and N2/H2 gasses to stabilize temperature and heat exchange. Before performing ReaxFF based simulations, the behaviour of the systems was investigated by means of first-principles MD simulations, as done in the case of bare silicon clusters, to test the effects of the different conditions on the reactivity of the system. Two different models were simulated.
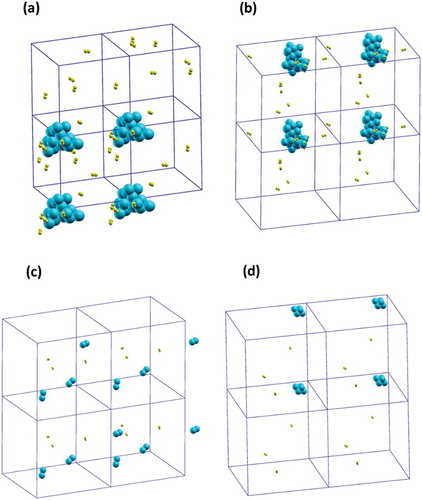
The first one consisted of a Si20 cluster and 10 H2 molecules at T = 2000 K in a cubic cell where the side was1.85 nm (the starting configuration is shown in )). In order to reproduce the experimental conditions (P = 1.00 atm), the unit cell side should have been approximately 15.5 nm. Considering that the calculations in such a cell would have been totally prohibitive with the computational resources at our disposal, we reduced the cell side to the value reported above. This determined a huge pressure increase (P ≈ 600 atm). Although, in principle, the high pressure could be considered unphysical and could enhance the dissociation of the hydrogen molecules on the silicon cluster due to both enthalpy (adsorption is exothermic) and entropy (reduction of translational free energy) contributions, it was found that in the explored time range (about 25 ps), no dissociation event of the H2 molecule over the cluster took place. The simulation time was extended to 50 ps and also in this case no H2 dissociation was observed. This is a first indication of a poor reactivity between the growing Si particles and the hydrogen atmosphere at the considered temperature. A further check was carried out by including a Si dimer in the cell to explore the reactive behaviour of small Si fragment towards H2 and Si20. The Si dimer spontaneously adsorbed onto the Si20 cluster surface. The cell volume was then increased by a factor of approximately 4, to reduce pressure (up to about 160 atm) and estimate its effect on the simulation results. In this case the dimer did not react immediately with the cluster surface but reacted, instead, with two H2 molecules forming a Si2H4 species in a time span of about 20 ps, and then it was adsorbed on the Si20 cluster, see ). These results pointed out the remarkable reactivity of small Si species with the molecular gas and suggested that it was necessary to simulate a huge variety of model systems containing small Si clusters to obtain a significant statistical sample able to depict possible behaviours of NPs growth in reactive gasses.
Figure 3. (a) Si20 surrounded by 10 H2 molecules; (b) Si20 surrounded by 10 H2 molecules and one Si dimer in its final configuration where the dimer has adsorbed on the bigger cluster in the form of Si2H4 species; starting (c) and final (d) configuration of 3 Si dimers surrounded by 3 H2 molecules: in the final configuration the three dimers have coalesced in a Si6 cluster. PBC cubic boxes are visualized to give an idea of the concentration of the species. Si and H atoms are cyan and yellow, respectively.
From the QC data collected, we could speculate that reactivity of large SiNPs in relation to the ambient gas is much smaller than that observed for small Si clusters; in fact, notwithstanding the very high pressure, H2 was not able to dissociate on the Si20 cluster. To further confirm this hypothesis, the reactivity of Si dimers in more realistic conditions was investigated in detail using a second model system.
The second model consisted of 3 Si2 and 3 H2 molecules at T = 2000 K in a cubic cell, where the side was 3.50 nm, at P ≈ 47 atm. The starting and final configurations are shown in ), respectively. The results of this simulation indicated that in a time span of about 25 ps the 3 Si dimers had coalesced (without impediments) forming a Si6 cluster. No dissociation of the H2 molecules took place. The high reactivity of the Si dimer in the former system was probably due to the high pressure of the reactive gas. In fact, when reducing the pressure, hydrogen-silicon reactivity was completely suppressed (in line with the experiments).
To explore the low reactivity of the H2 gas during SiNPs growth that resulted from QCMD simulations in a more realistic environment, two different systems were investigated at the classical level by using the Si force field parameters developed previously [Citation22,Citation23] and Si-H and Si-N parameters, found in Ref [Citation13,Citation36]., respectively. Briefly, the systems consisted of the same Si cluster of 1.7 nm in radius used in the sintering simulations (made by about 1000 Si atoms) and 187 H2 molecules or 187 Si atoms at T = 2000 K in a cubic cell where the side was 40.0 nm, at P = 1.00 atm. The choice of considering two models, namely silicon cluster surrounded by either H2 molecules or Si atoms, had the purpose to compare particle growth and H2 dissociation on the nanoparticle surface. The final results perfectly agree with the indications of the QMD simulations: during the whole simulation time (1 ns): in the first model only one out of 187 H2 molecules dissociated, see ), whereas, in the other case about 18 Si atoms were found attached to the cluster (not shown).
Figure 4. (a-b) Si particle made of 1000 atoms surrounded by 187 H2 molecules; after 1 ns only one molecule was physisorbed and two H atoms chemisorbed on the cluster surface; (c-d) Si particle made of about 100 atoms surrounded by 30 H2 and 30 N2 molecules; after 2 ns the particle was covered with six chemisorbed hydrogens but no N atoms were found on the surface. Si, H and N atoms are cyan, yellow and blue, respectively.
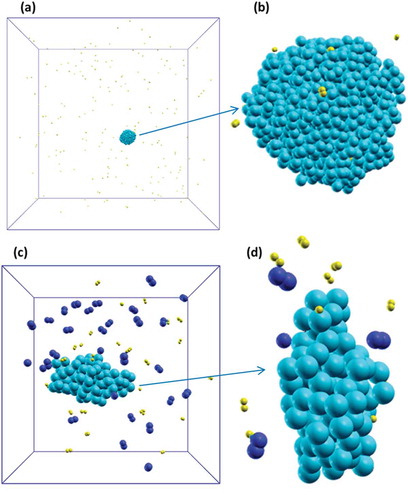
Moving to the reactive behaviour of Si species when both H2 and N2 molecules were present, it was found that N2 was less reactive than H2 (trial calculations at the QCMD level). This was essentially due to the fact that N2 had a higher binding energy to the surface. To verify this assumption, we considered a system with a cluster made by about 100 atoms surrounded by 30 H2 molecules and by 30 N2 molecules at T = 2000 K in a cubic cell where the side was 5.0 nm; the final configuration is shown in . At the end of the simulation (after 2 ns), the cluster was covered with 6 chemisorbed H but no N atoms were found on the surface. As already observed in the other cases examined in this work, reactivity of H2 was enhanced by the high pressure inside the cell. This, however, was not sufficient to trigger the dissociation of the N2 molecules, which remained intact around the Si cluster sometimes interacting with its surface, confirming the prediction of an extremely low reactivity at the considered temperature and on the time scale of the Si particles growth.
To evaluate further the behaviour of H2 and N2 on silicon clusters their dissociation barriers were calculated by means of metadynamics [Citation37,Citation38] ().
The free energy profiles displayed in depict the behaviour of a system made of a H2 or N2 molecule and a Si20 cluster in a cubic cell where the side is 2.0 nm (pressure about 30 atm). Total sampling time and temperature were around 2 ns and 1000 K, respectively. To reduce the computational efforts related to the sampling, energies and forces were calculated at the DFTB level by using the algorithm encoded into the CP2K package.
Figure 5. Left panel: DFTB free energy surface of the Si20 + H2 system. Right panel: DFTB free energy surface of the Si20 + N2 system.
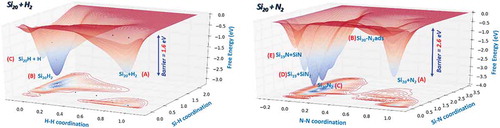
The left panel in shows the free energy surface as a function of the H-H and Si-H distances (coordinations in ). The main three minima, identified by the calculations, are indicated with the letters in parenthesis: A is the starting point of the simulation (i.e H2 and Si20 in the gas phase); B is the deepest minimum corresponding to the H2 dissociation where the two hydrogens are separately adsorbed on the Si20 cluster; C is related to the H2 dissociation with one hydrogen adsorbed on the cluster and the other one free in the gas phase. The blue dots indicate the minimum free energy path to the hydrogen dissociation and the calculated barrier is about 1.6 eV. At atmospheric pressure this value had to be corrected by adding 0.4 eV, which corresponds to an entropic contribution related to the pressure reduction. Finally, the dissociation rate estimated for H2 through the transition state theory was approximately 2.6 ns. The dissociation free energy barrier of N2 was found larger than the one identified in the case of the hydrogen molecule. The free energy landscape of the Si20 + N2 system shown in the right panel of reveals more complex trends than the one corresponding to H2 and many basins are identified: A is the initial state with N2 and Si20, separated in the gas phase, B corresponds to the undissociated N2 molecule absorbed on the Si20 cluster; C are the two deepest minima where the N2 molecule is dissociated. In this case both nitrogen atoms are connected to the surface and present a different degree of penetration. Minima D and E are the dissociated states of N2 that have left the cluster surface as NSiN (D) or SiN (E). A careful examination of the dissociation mechanism revealed that it was a two-step process in which the N2 molecule first adsorbed molecularly on the surface cluster and then dissociated. The barrier for going from A to C is around 2.6 eV, which corresponds to a N2 dissociation rate of approximately 800 ns in the plasma conditions.
These results suggest that both H2 and N2 reactive gasses can influence the nanoparticle generation mechanism by effective collisions with the Si clusters present in the plasma. Reactions involving nitrogen seem less probable than effective interactions with hydrogen, which showed also a slight propensity to penetrate into Si configurations.
4. Conclusions
In this paper, we have described a computational protocol aimed at investigating the process of growth of SiNPs in plasma conditions by exploiting a reactive force field (ReaxFF), calibrated and validated on accurate first-principles methods. The use of plasma reactors is technologically relevant as it does not employ chemical solvents, but lacks fine control of size distribution and composition of the produced NPs. Modelling plays a major role in unveiling the complex and interwoven processes which take place in a realistic reactor and a multi-scale approach becomes a fundamental tool to simultaneously guarantee a reliable level of accuracy and significant statistical sampling. The protocol used here has been designed for silicon but can be extended to other materials grown via plasma reactors, as in the case of zinc oxide [Citation29].
In the investigation of the growth process, we have shown that the concept of critical size in a nucleation stage loses its validity in the atomistic perspective, where already a silicon dimer is a stable species. Thus, we have introduced the concept of inception time (as a function of size, temperature and pressure) to describe the formation of SiNPs. Moving from small clusters to larger NPs, we have shown how a growth mechanism due to the encounter of two ‘big’ particles becomes important. We have investigated in detail the process of coalescence/sintering between SiNPs, showing that a major role is played by the physical state (either solid or liquid) of NPs. We have monitored the sintering process and proposed an efficient scheme to investigate finite particles behaviour, including melting.
To consider more realistic gas mixtures of typical microwave low-temperature plasma, we have disclosed how the presence of H2/N2 contaminants influences nucleation and growth, by means of a combination of first-principles and ReaxFF dynamics. Reactivity of small Si clusters with hydrogen forming silane-like species could be induced at high temperature only in case of very high pressure, quite far from those characterizing typical low-temperature plasma reactors. When shifting to realistic conditions, in fact, silicon-hydrogen reactivity is suppressed and the presence of hydrogen molecules in the gas mixture does not play any relevant role. Due to its high stability, the nitrogen molecules do not react in any of the considered conditions of temperature and pressure.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Acknowledgments
This work has been developed as part of the European project Nanodome that has received funding from the European Union’s Horizon 2020 Research and Innovation Programme, under Grant Agreement number 646121. The authors thank A.C.T Van Duin for providing the serial version of the ReaxFF program and thank the SCM experienced team for their excellent technical support and suggestions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Leach AR. Molecular modelling : principles and applications. Pearson Education; 2 edition (2001), Harlow, England.
- Van Duin ACT, Dasgupta S, Lorant F, et al. ReaxFF: A reactive force field for hydrocarbons. J Phys Chem A. 2001;105:490–506.
- Senftle TP, Hong S, Islam MM, et al. The ReaxFF reactive force-field: development, applications and future directions. Npj Comput Mater. 2016;2:15011.
- Goddard WA, Van Duin ACT, Strachan A, et al. ReaxFFSiO reactive force field for silicon and silicon oxide systems. J Phys Chem A. 2003;107:3803–3811.
- Buehler MJ, van Duin ACT, Goddard WA. Multiparadigm modeling of dynamical crack propagation in silicon using a reactive force field. Phys Rev Lett. 2006;96:095505.
- Buehler MJ, Tang H, van Duin ACT, et al. Threshold crack speed controls dynamical fracture of silicon single crystals. Phys Rev Lett. 2007;99:165502.
- Naserifar S, Liu L, Goddard WA, et al. Toward a process-based molecular model of SiC membranes. 1. Development of a reactive force field. J Phys Chem C. 2013;117:3308–3319.
- Jaramillo-Botero A, Naserifar S, Goddard WA. General multiobjective force field optimization framework, with application to reactive force fields for silicon carbide. J Chem Theory Comput. 2014;10:1426–1439.
- Motevalian SP, Aro SC, Cheng HY, et al. Kinetics of silane decomposition in high-pressure confined chemical vapor deposition of hydrogenated amorphous silicon. Ind Eng Chem Res. 2017;56:14995–15000.
- Hahn SH, Rimsza J, Criscenti L, et al. Development of a ReaxFF reactive force field for NaSiO x/water systems and its application to sodium and proton self-diffusion. J Phys Chem C. 2018;122:19613–19624.
- Krishnan NMA, Wang B, Sant G, et al. Revealing the effect of irradiation on cement hydrates: evidence of a topological self-organization. ACS Appl Mater Interfaces. 2017;9:32377–32385.
- Soria FA, Zhang W, van Duin ACT, et al. Thermal stability of organic monolayers grafted to Si(111): insights from ReaxFF reactive molecular dynamics simulations. ACS Appl Mater Interfaces. 2017;9:30969–30981.
- Soria FA, Zhang W, Paredes-Olivera PA, et al. Si/C/H ReaxFF reactive potential for silicon surfaces grafted with organic molecules. J Phys Chem C. 2018;122:23515–23527.
- https://www.scm.com. (2017)
- Münzer A, Sellmann J, Fortugno P, et al. Inline coating of silicon nanoparticles in a plasma reactor: reactor design, simulation and experiment. Mater Today Proc. 2017;4:S118–S127.
- Harry JE. Introduction to Plasma Technology. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2010.
- Mangolini L. Synthesis, properties, and applications of silicon nanocrystals, J. Vac. Sci. Technol. B. Nanotechnol Microelectron Mater Process Meas Phenom. 2013;31:020801.
- Priolo F, Gregorkiewicz T, Galli M, et al. Silicon nanostructures for photonics and photovoltaics. Nat Nanotechnol. 2014;9:19–32.
- Giannozzi P, Baroni S, Bonini N, et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J Phys Condens Matter. 2009;21:395502
- Rossi G, Ferrando R. Searching for low-energy structures of nanoparticles: a comparison of different methods and algorithms. J Phys Condens Matter. 2009;21:084208.
- Barcaro G, Fortunelli A. A magic Pd−Ag binary cluster on the F s -defected MgO(100) surface. J Phys Chem C. 2007;111:11384–11389.
- Barcaro G, Monti S, Sementa L, et al. Atomistic modelling of Si nanoparticles synthesis. Crystals. 2017;7:54.
- Barcaro G, Monti S, Sementa L, et al. Parametrization of a reactive force field (ReaxFF) for molecular dynamics simulations of Si nanoparticles. J Chem Theory Comput. 2017;13:3854–3861.
- van Duin ACT, Baas JMA, van de Graaf B. Delft molecular mechanics: a new approach to hydrocarbon force fields. Inclusion of a geometry-dependent charge calculation. J Chem Soc Faraday Trans. 1994;90:2881.
- Iype E, Hütter M, Jansen APJ, et al. Parameterization of a reactive force field using a Monte Carlo algorithm. J Comput Chem. 2013;34:1143–1154.
- ADF2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available from: http://www.scm.com. 2017
- A. C. T. Van Duin, ReaxFF User Manual. ReaxFF User Man., 2002, 1–39.
- LAMMPS Molecular Dynamics Simulator. [accessed on 2017 Feb 12]. Available online: http://lammps.sandia.gov, http://lammps.sandia.gov.
- Barcaro G, Monti S, Sementa L, et al. Modelling nucleation and growth of ZnO nanoparticles in a low temperature plasma by reactive dynamics. J Chem Theory Comput. 2019;153:2010–2021
- Karthika S, Radhakrishnan TK, Kalaichelvi P. A review of classical and nonclassical nucleation theories. Cryst Growth Des. 2016;16:6663–6681.
- Zachariah MR, Carrier MJ. Gas-phase nanoparticle sintering : a comparison with phenomenological models. J. Aerosol. Sci.1999;30:1139–1151.
- Di Nunzio PE, Martelli S. Coagulation and aggregation model of silicon nanoparticles from laser pyrolysis. Aerosol Sci Technol. 2006;40:724–734.
- Sementa L, Barcaro G, Monti S, et al. Molecular dynamics simulations of melting and sintering of Si nanoparticles: a comparison of different force fields and computational models. Phys Chem Chem Phys. 2018;20:1707–1715.
- Baletto F, Ferrando R. Structural properties of nanoclusters: energetic, thermodynamic, and kinetic effects. Rev Mod Phys. 2005;77:371–423.
- Chau J, Yang -C-C, Shih -H-H, et al. Microwave plasma production of metal nanopowders. Inorganics. 2014;2:278–290.
- Zhou T, Liu L, Goddard WA III, et al. ReaxFF reactive molecular dynamics on silicon pentaerythritol tetranitrate crystal validates the mechanism for the colossal sensitivity. Phys Chem Chem Phys. 2014;16:23779–23791.
- Barducci A, Bonomi M, Parrinello M. Metadynamics. Wiley Interdiscip Rev Comput Mol Sci. 2011;1:826–843.
- Sutto L, Marsili S, Gervasio FL. New advances in metadynamics. Wiley Interdiscip Rev Comput Mol Sci. 2012;2:771–779.