
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Peripheral membrane proteins (PMPs) bind temporarily to the surface of biological membranes. They also exist in a soluble form and their tertiary structure is often known. Yet, their membrane-bound form and their interfacial-binding site with membrane lipids remain difficult to observe directly. Their binding and unbinding mechanism, the conformational changes of the PMPs and their influence on the membrane structure are notoriously challenging to study experimentally. Molecular dynamics simulations are particularly useful to fill some knowledge-gaps and provide hypothesis that can be experimentally challenged to further our understanding of PMP-membrane recognition. Because of the time-scales of PMP-membrane binding events and the computational costs associated with molecular dynamics simulations, membrane models at different levels of resolution are used and often combined in multiscale simulation strategies. We here review membrane models belonging to three classes: atomistic, coarse-grained and implicit. Differences between models are rooted in the underlying theories and the reference data they are parameterized against. The choice of membrane model should therefore not only be guided by its computational efficiency. The range of applications of each model is discussed and illustrated using examples from the literature.
Graphical abstract
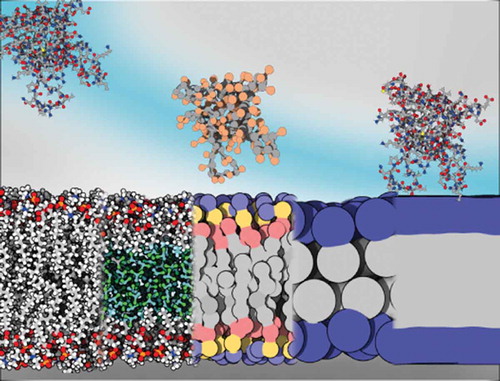
1. Introduction
Peripheral membrane proteins (PMP) are soluble proteins that bind transiently to the surface of biological membranes () where they accomplish their functions [Citation1]. Unlike transmembrane (TM) proteins, they do not span the entire lipid bilayer but interact with the membrane through a collection of protruding hydrophobic residues located on loops or amphipathic helices [Citation2–6] which, according to the text-book model, are often surrounded by positively charged amino acids lysines and arginines [Citation7]. The term peripheral protein encompasses proteins performing various critical functions including enzymes involved in lipid metabolism or blood coagulation, membrane remodelling machines such as BAR domains [Citation8] or ESCRTIII [Citation9], lipid-transfer proteins and well-known membrane-targeting domains such as C1, C2, FYVE, PH, PX, ENTH and GLA [Citation10,Citation11], to name a few.
Figure 1. Membrane models and their respective compatible protein description. Membranes from left to right: all-atom, HMMM, MARTINI, SBCG, implicit. Proteins (Pleckstrin Homology (PH) domain, 2DA0) from left to right: all-atom, all-atom, half-MARTINI/half-RBCG, SBCG, all-atom. Rendered with 3DProteinImaging [Citation12]
![Figure 1. Membrane models and their respective compatible protein description. Membranes from left to right: all-atom, HMMM, MARTINI, SBCG, implicit. Proteins (Pleckstrin Homology (PH) domain, 2DA0) from left to right: all-atom, all-atom, half-MARTINI/half-RBCG, SBCG, all-atom. Rendered with 3DProteinImaging [Citation12]](/cms/asset/75dadb55-bfab-409f-b071-548e56a5246f/tapx_a_1932589_f0001_oc.jpg)
Peripheral proteins bind biological membranes with an exquisite resolution in time and space, often thanks to a fine-tuned lipid specificity achieved using amino acid motifs exposed on their surface. The residence times of peripheral proteins vary depending on their function, and for example the residence time of a bacterial phospholipase C on small unilamellar vesicles was estimated to 379 ± 49 ms, in agreement with earlier estimations for a highly related PLC [Citation13] or for human phospholipase Cγ2 [Citation14]. The turn-over rates for membrane-associated proteins in clathrin-mediated endocytosis have been estimated to a few seconds [Citation15]. Accordingly, PMPs bind membrane through different mechanisms. Some bind a lipid head to a deep pocket, some bind thanks to a covalent lipid anchor and some have neither, relying on displaying on their surface the necessary structural and sequence motif to bind to membrane lipids. In this review, the region interacting with the membrane lipids is referred to as the interfacial-binding site or IBS. The associated amino acids motifs on the surface of membrane-targeting domains have been the subject of numerous studies [Citation11] but other PMPs are not that well characterized. Besides phosphoinositides a number of other lipids are also recognized by membrane targeting domains (and other PMPs) but we are only starting to grasp the chemical complexity of these interactions as the complexity of biological membranes is slowly uncovered [Citation16,Citation17].
Because they most often exist in a soluble form, obtaining structures of PMPs has historically been easier than for TM proteins. While progresses in structure determination of TM proteins has led to a rapid increase of the number of structures available in public databases, the number of structures of proteins with weaker membrane association is comparatively small [Citation18]. This applies to bitopic and monotopic TM proteins, and to PMPs. Due to the transient nature of their interaction to membranes, most structural biology methods are excluded as a direct source of information, and we lack information on the location of the IBS, and about potential conformational changes happening upon membrane association for example. Moreover, the IBSs of peripheral proteins are more challenging to predict from sequence or structure than transmembrane protein segments. The structure and amino acid composition of these interfaces is not as characteristic as those of transmembrane regions, which often consist in helical bundles enriched in hydrophobic residues, making them easily predictable from amino acid sequence only [Citation19]. The prototypical IBS is described as a combination of lysines and arginines and only a few hydrophobic amino acids [Citation1]. Peripheral proteins are indeed restricted in terms of the hydrophobic patches they can expose on their surface and still retain solubility. Such a mix of polar and hydrophobic residues on the surface of the protein is not easy to discriminate from the rest of the protein surface and even less so, from the primary sequence. Moreover, even if the role of unspecific electrostatic interactions has been demonstrated for several protein families [Citation7,Citation20], recent studies report more complex and subtle recognition mechanisms for some proteins. That includes weak electrostatic interactions, cation-pi interactions () [Citation3,Citation21,Citation22] between aromatic amino acids and phosphatidylcholine-containing lipids, roles of ions in mediating lipid–protein interactions [Citation23], or the importance of macrodipoles [Citation20,Citation24,Citation25]. In summary, while we have high quality structural data of some peripheral proteins in their soluble forms, their IBS is seldomly clearly identified and their binding mechanism remain elusive.
Figure 2. Tyrosine-choline π-cation interactions in Bacillus thurigiensis phosphatidylinositol-specific phospholipase C anchored at the surface of a PC lipid bilayer. Reprinted from [Citation3], with permission from ACS Publications
![Figure 2. Tyrosine-choline π-cation interactions in Bacillus thurigiensis phosphatidylinositol-specific phospholipase C anchored at the surface of a PC lipid bilayer. Reprinted from [Citation3], with permission from ACS Publications](/cms/asset/1c7ec861-4119-4255-ac78-078199494ffb/tapx_a_1932589_f0002_oc.jpg)
Molecular dynamics (MD) simulations have proven useful in predicting IBS of PMPs and modelling PMP binding to lipid bilayers [Citation26–28]. MD simulations simulate the behavior of a molecular system by solving the Newton’s equation of motion for each particle constituting the system. The system simulated usually consists of several molecules and in MD simulations of PMPs, it is natural to include at least the protein itself, a bilayer formed by lipids relevant to the cellular membrane being modeled, water molecules solvating both sides of the membrane and relevant ions. For a regular size enzyme like a phospholipase D from recluse spider venom (285 amino acids) that we recently simulated, 256 POPC lipids and 22,613 water molecules were needed plus 4 ions, so a system totaling nearly 110,000 atoms.
Interactions between particles are described using a so-called force field. In molecular mechanics (MM) atomistic force fields, each atom is represented by a single particle carrying a point charge, following the Born Oppenheimer approximation. As we will see in this review, there also exists force fields using a coarser description of the simulated molecules where a group of atoms can be represented by a single particle for the sake of the simulation.
A generic atomistic FF for biomolecules expresses the potential of the system as a sum of bonded terms and non-bonded terms. Bonded terms include harmonic potentials for bond stretching and valence angle bending, and a cosine function for torsions. Non-bounded interactions are accounted for by pairwise terms; a coulombic potential for interactions between point charges and a Lennard Jones potential for van der Waals interactions (Cf Equationequation 1)1
1 .
Non-bounded interactions are the computational bottleneck in MD simulations, as they scale in principle as a function of N2, where N is the number of particles simulated. In practice, the use of cut-off schemes and neighbour lists reduces the number of pairwise interactions to be estimated at each integration step and decreases the computational cost associated with the calculation of the coulombic and Lennard Jones terms. Even though MD simulations remain expensive for large systems and on long timescales, such as PMP association to -or desorption from- a membrane. For the 110,000 atoms system described above, using NAMD (v 2.13) [Citation29] and the CHARMM36 force field [Citation30–32], we simulated 100 nanoseconds per day on 1024 cores of the newest high-performance computer in Norway at the time of writing (Betzy: BullSequana XH2000, ranked 61st on the top500 list in November 2020Footnote1).
A number of schemes exist to more efficiently explore the phase space; enhanced sampling and biasing methods such as replica exchange MD [Citation33], accelerated MD [Citation34], umbrella sampling [Citation35] and steered MD [Citation36] to name a few. In steered MD for example, an external force, represented by a new term in the potential, is applied to a group of atoms.
Atomistic MD simulations are particularly useful to describe interactions between the protein residues and the interfacial lipid groups [Citation2,Citation3,Citation37–40]. Moreover, the improvements brought to atomistic force fields in the last decade have among enabled simulations of a wider array of lipids [Citation41]. Yet capturing the spontaneous membrane association of some PMPs is elusive with conventional atomistic MD simulations. One of the reasons is the slow lateral lipid diffusion (D) which is typically on the order of 10−8 cm2.s−1 at ambient temperature [Citation39,Citation42]. Therefore, during a regular atomistic MD simulation (hundreds of nanosecond to microseconds), the bilayer may appear relatively static preventing efficient sampling of lipid–PMP interactions. In order to overcome this problem, different membrane models can be used. There exist indeed a variety of membrane models amenable to simulations. It is common to use a multiscale strategy taking advantage of the computational efficiency of the earliest membrane models, and of the more recent improved accuracy of lipids force fields. The first membrane model we are aware of dates back to 1988 and was used by Edholm and Jähnig to simulate a transmembrane helix from glycophorin [Citation43]. They could simulate one time-step in 0.3 s on a CRAY-1 supercomputer. That model and the following ones represent the membrane as a hydrophobic slab with a polar layer on either sides and are meant to compute the energetic of solvation of membrane proteins in or at the surface of membranes. In essence, coarse-grained (CG) models are somewhat related to the spirit of the implicit models as they too are parameterized against partition coefficients, but CG models present the clear advantage of treating the lipids as individual molecules. CG models are computationally efficient but lack the ability to describe the fine details of protein–lipid interactions that might be needed to explain selective protein-membrane binding. For this, one needs atomistic force fields [Citation41,Citation44].
Biological membranes consist mainly of glycerophospholipids but contain also other molecules, including cholesterol, proteins, etc. [Citation45]. Lipid bilayers form a complex chemical environment, with a hydrophobic core and a polar interfacial region, and their physico-chemical and mechanical properties influence, and are influenced by the membrane-associated proteins. Accounting for the chemical complexity of the interfacial region, lipid diffusion, lateral pressure, curvature stress, electrostatic properties (transmembrane, surface and dipole potentials) and more, is extremely challenging for models that also need to be computationally efficient. In what follows we describe membrane models relevant for simulations of peripheral membrane proteins and how the different levels of resolution of these models are suited to address different aspects of the mechanism of peripheral membrane binding of proteins. We illustrate the use of these methods with a number of selected examples from which peptides and transmembrane proteins are excluded. The latter have been extensively reviewed elsewhere recently [Citation27,Citation46,Citation47].
2. Membrane models for molecular dynamics simulations
2.1 Atomistic models
The main atomistic force fields for MD simulations of lipid bilayers are CHARMM, AMBER, OPLS-AA, Slipids, and GROMOS. They are derived from atomistic force fields for proteins and other biomolecules and represent lipid molecules in all-atom (AA) detail or in a united-atom (UA) fashion where hydrogens on non-polar chemical groups are not explicitly represented, thus speeding up the simulations. The reader is referred to e.g. Refs. [Citation41,Citation44] and references therein for a detailed account of the development efforts. Here, we briefly overview the most popular AA and UA force fields [Citation48], including the atomistic Highly Mobile Membrane-Mimetic (HMMM) [Citation49].
2.1.1 All-atoms force fields
The CHARMM (Chemistry at HARvard Macromolecular Mechanics) force field for lipids is widely used in simulations of lipid bilayers, and compatible with the CHARMM force field for proteins [Citation31,Citation32,Citation50]. CHARMM describes hydrogen atoms explicitly. The CHARMM force field for lipids has been continuously improved from the initial CHARMM22 lipid parameter set [Citation51,Citation52] developed in the 1990s to the most recent update CHARMM36 [Citation30]. Briefly CHARMM27 [Citation53] and CHARMM27r [Citation54,Citation55] suffered from a number of limitations, including too high torsional barriers along the lipid tails (C27), overstabilization of gauche states (C27), and the tendency of saturated phosphatidylcholine bilayers to transition into the gel phase above the experimental phase transition temperature (C27r). This could be avoided by applying surface tension. Extensive parameterization efforts of the lipid parameter set have led to CHARMM36 which can be used without the need for an external surface tension, and faithfully reproduce the physico-chemical properties of a wide variety of lipid bilayers including glycerophospholipids (saturated, unsaturated, branched, containing cycles) with various headgroups (phosphatidylcholine PC, phosphatidylacid PA, phosphatidylglycerols PG, phosphatidylinositol PI, phosphatidylserine PS), glycolipids, lipopolysaccharides, sphingolipids, ceramides, and a range of sterols. Noteworthy are also our efforts to improve the description of cation-π interactions between choline heads and aromatic amino acids [Citation56,Citation57] observed between PC lipids and tyrosine or tryptophane residues of several peripheral protein [Citation2,Citation3,Citation21,Citation58,Citation59]; modifications of the CHARMM36 Lennard-Jones potential between choline and aromatics resulted in a better agreement for cation-pi interactions (see ). The parameters are available in the CHARMM-WYF parameter set.
Figure 3. Phenol-tetramethylammonium potential energy surface obtained with quantum mechanics calculations, the CHARMM36 force field and the CHARMM-WYF modification of the Lennard-Jones potential. Reprinted from [Citation56], with permission from ACS Publications
![Figure 3. Phenol-tetramethylammonium potential energy surface obtained with quantum mechanics calculations, the CHARMM36 force field and the CHARMM-WYF modification of the Lennard-Jones potential. Reprinted from [Citation56], with permission from ACS Publications](/cms/asset/1d768f46-6681-495c-a7c8-95d6b89fc10a/tapx_a_1932589_f0003_oc.jpg)
AMBER (Assisted Model Building with Energy Refinement), like CHARMM, treats all hydrogen atoms explicitly [Citation60]. Compared to the CHARMM force fields, AMBER force fields were generally less used for membrane protein simulations due to the lack of a specific parameter set for lipids [Citation61]. The general AMBER force field, known as GAFF (Generalized Amber Force Field) [Citation62] and meant for simulation of arbitrary organic molecules was extended to include lipid parameters in 2007 [Citation63]. This parameter set suffered from some of the same limitations as the CHARMM27 parameter set. In 2012, two somewhat improved force fields were released; GAFFlipid [Citation64] and Lipid11 [Citation65] (Lipid11 was released with AMBER12). Subsequent efforts focusing on head and tail groups charges, Lennard-Jones and dihedral angles parameters resulted in 2014 in Lipid14 [Citation66] a modular lipid force field for tensionless lipid phospholipid simulations and compatible with the AMBER protein force field. Slipids [Citation67,Citation68] (Stockholm Lipids) is another force field compatible with AMBER, but it originates from efforts [Citation69,Citation70] to improve the CHARMM27r parameter set. The Slipids force field is available for saturated and unsaturated glycerophospholipids (PC, PS, PE, PG), sphingolipids (sphingomyelin), and cholesterol.
The OPLS-AA (Optimized Parameters for Liquid Simulations All Atom) [Citation71] force field was developed with emphasis on reproducing the experimental thermodynamical and partitioning properties of short chain hydrocarbons alkanes, and now includes parameters for several classes of biomolecules. It was recently extended to better model long chain hydrocarbons [Citation72], and lead to a parameter set called L-OPLS that has expanded its repertoire of lipids to include PC, PE, unsaturated lipids and cholesterol [Citation73], as well as plasmalogens and PE headgroups [Citation74]. Yet, so far, the availability of lipids in the L-OPLS force field has not been as diverse as that of the CHARMM36 and the AMBER-compatible force fields.
2.1.2 United-atoms force fields
Compared to the AA force fields for lipids, the united-atom model claimed to provide a better balance between accuracy and computational cost in membrane modelling [Citation48]. Among currently available UA lipid force fields, GROMOS (GROningen MOlecular Simulation package) [Citation75–77] has the most popular ones and offers the largest variety of lipids. In the GROMOS force fields, nonpolar CH, CH2, and CH3 groups of hydrocarbons are considered as a single particle. The protein force field is parameterized against thermodynamic properties of aliphatic chains to enthalpies and free energies of solvation in polar and apolar environments [Citation78]. There are two main classes of GROMOS force field: those with the original GROMOS non-bonded parameters [Citation77,Citation79] and the ones with Berger modifications [Citation80,Citation81]. In the Berger modifications, in addition to the modification of non-bonded interaction parameters, a Ryckaert-Bellemans potential (cosn(ψ)) is used to describe torsions of the hydrocarbon chains [Citation77,Citation82]. The Berger lipid parameters are recommended to maximize the lipid bilayer sampling due to its fast diffusion and good simulation efficiency [Citation83]. Although Berger lipid is one of the most popular lipid force fields the combination of Berger/GROMOS force fields overestimates the protein lipid interactions and consequently results in drastic changes of lipid properties upon protein insertion [Citation84].
2.1.3 Highly Mobile Membrane-Mimetic Model
Figure 4. Regular atomistic bilayer (right) and HMMM bilayer (left) model. Figure inspired by the Ohkubo et al. 2012 [Citation49] and Vermaas et al. 2015 [Citation85]
![Figure 4. Regular atomistic bilayer (right) and HMMM bilayer (left) model. Figure inspired by the Ohkubo et al. 2012 [Citation49] and Vermaas et al. 2015 [Citation85]](/cms/asset/792672e3-b5b7-4141-843e-7e97d3e866bd/tapx_a_1932589_f0004_oc.jpg)
HMMM [Citation49,Citation86] uses an AA force field, namely CHARMM36 [Citation30], but represents the lipid bilayer with short-tailed (ST) surfactant-like lipids and a liquid phase for the membrane core, thus increasing lateral lipid diffusion.
In 2011, Arcario et al., used a biphasic solvent model to simulate the membrane insertion of a coagulation protein, namely the GLA domain of human protein C [Citation86]. MD simulations with a full tail (FT) atomistic lipid bilayer did not lead to spontaneous binding of the GLA domain, and one had to resort to steered MD [Citation36] for the protein to anchor at the bilayer interface [Citation87]. With the biphasic solvent model though, the protein did bind the bilayer spontaneously within the early nanoseconds of simulation at a similar depth of anchoring and orientation similar to that obtained with the enhanced sampling simulation. In this model, the lipid bilayer was replaced by a single layer of 1,1-dichloroethane (DCLE).
In 2012, the biphasic solvent was enhanced by adding two extra layers of surfactant-like ST lipids on each side of the organic solvent phase () [Citation49]. In this updated HMMM membrane representation, the bilayer core is represented by a DCLE layer, and the surfactant-like molecule is divalerylphosphatidylserine (DVPS), a saturated ST lipid with five acyl tail carbons. A mixture of DVPS with DCLE forms a bilayer-like structure within 20 ns in a MD simulation, and the lateral diffusion of this membrane model is five times higher than a FT model composed of 1,2-dioleoyl-sn-glycero-3-phospho-L-serine. The performance of the HMMM representation to model the spontaneous insertion of a PMP was assessed by 10 simulations (replicas) of a GLA domain that all converge toward the conformation observed in steered MD [Citation87].
The permeability of the HMMM membrane was validated using the degree of hydration and ions penetration and showed good agreement with AA FT lipid bilayers [Citation49]. Using potential of mean force (PMF) calculations, Pogorelov et al [Citation88]. assessed the free energy of transfer of sidechain analogs of ten selected amino acids (I, A, Y, W, E, N, S, C, D and K) to a HMMM membrane. They reported good agreement with existing experimental, atomistic and coarse-grained free energy profiles although they did highlight some differences. First, D was slightly more stable in the DCLE layer than it would be between FT lipid tails. Aromatic residues (F, Y and W) diverge from the reference in the tail and DCLE region and especially so for TRP; their stability in the membrane core is overestimated by the HMMM PMFs. Most noticeably, HMMM fails to capture the minimum in the interfacial region with positive PMFs for ASN and SER, while the CYS analog has its minimum in the membrane core. The deviations from reference PMFs in the core region are likely due to two factors: the polar nature of DCLE leading to dipole-dipole interactions with polar groups and its fluidity allowing sidechain tumbling in the bilayer core.
HMMM was also used to characterize the membrane-binding site of the catalytic domain of cytochrome P450 3A4 (CYP3A4) [Citation89], an enzyme well-known for its involvement in xenobiotics and endogenous compound metabolism [Citation90,Citation91]. The simulations revealed reorientations of side chains upon membrane binding, which promote the opening of access tunnels leading to the active site of CYP3A4
Naturally simulations with HMMM cannot account for effects due to the nature of lipid tails, differences between e.g. phospholipids and sphingolipids, or the presence of sterols. Also, the HMMM membrane model is not meant to reproduce mechanical properties (compressibility or bending) of lipid bilayers. The HMMM builder [Citation92,Citation93] in the CHARMM-GUI [Citation94] can be used to generate simulation input files with the HMMM model.
2.1.4 All-atoms/united-atoms MD simulations of lipid transfer proteins
Figure 5. Schematic atomistic representation of the α-tocopherol transfer protein interacting with a DOPC/DOPE phospholipid bilayer (grey). The mobile gate (purple) at the entrance of the cavity interacts with the PIP2 headgroup (red) and the lipids. The neighbouring motif is coloured in blue, and the α-tocopherol molecule bound to the protein in yellow. The head of PIP2 facilitates anchoring of α-TTP to the membrane. Rendered with 3DProteinImaging [Citation12]. PDB ID: 3W67 [Citation95]
![Figure 5. Schematic atomistic representation of the α-tocopherol transfer protein interacting with a DOPC/DOPE phospholipid bilayer (grey). The mobile gate (purple) at the entrance of the cavity interacts with the PIP2 headgroup (red) and the lipids. The neighbouring motif is coloured in blue, and the α-tocopherol molecule bound to the protein in yellow. The head of PIP2 facilitates anchoring of α-TTP to the membrane. Rendered with 3DProteinImaging [Citation12]. PDB ID: 3W67 [Citation95]](/cms/asset/0028204e-a25e-4f5b-8e0c-8ca1c8189e19/tapx_a_1932589_f0005_oc.jpg)
The yeast Osh4 proteins are members of a family of lipid transfer proteins (LTPs) with oxysterol-binding protein (OSBP)-related domain (ORDs) as their lipid transfer domains. Osh4 transfer cholesterol from the endoplasmic reticulum (ER) to the plasma membrane (PM) in yeast cells. The atomistic details of membrane-binding mechanism of Osh4 to charged and uncharged lipid membrane models have been characterized computationally [Citation26,Citation96]. AA MD simulations using CHARMM36 force field on an Anton supercomputer [Citation97] showed that Osh4 binds strongly to anionic membranes in a single binding conformation. The binding event involved contact and interaction from six regions of the protein to the membrane. On the contrary, the Osh4 binds weakly and transiently to uncharged membranes with zwitterionic lipids and consequently does not form a stable binding conformation [Citation26]. The studies also revealed that the phenylalanine loop is one of the most important binding regions of Osh4 stabilizing the protein–membrane interaction [Citation26,Citation96]. Interestingly, an AA MD simulation on the oxysterol-binding protein-related protein, Osh6p, also showed that a corresponding loop in Osh6p has a similar critical role for anchoring the protein to anionic membrane. The Osh6p is a yeast LTP that transfers PS from the ER to the PM via PS/phosphatidylinositol 4-phosphate (PI4P) exchange cycles [Citation98]. The Osh6p is a yeast LTP that transfers PS from the ER to the PM via PS/phosphatidylinositol 4-phosphate (PI4P) exchange cycles. The study used the CHARMM36 force field and the GROMACS MD simulation engine and showed that Osh6p reduces its avidity for anionic membranes once it captures PS or PI4P in the lipid-binding pocket. The authors proposed an electrostatic switching mechanism that controls the lipid transfer activity of Osh6p between the ER and the PM.
Cholesteryl ester transfer protein (CETP) is another important transfer protein responsible for transferring cholesteryl esters (CE), triglycerides, and phospholipids (PLs) from high-density lipoproteins (HDL) to low-density lipoproteins and very low-density lipoproteins. A multiscale AA and CG MD simulation study of a CETP-lipoprotein complex revealed that CETP binds to the surface of HDL-like lipid droplets through its charged amino acids and tryptophan residues [Citation99]. The binding induces the formation of a small hydrophobic patch on the surface of the droplet and opens a route from the core of the CE lipid droplet to the binding pocket in CETP. This route is caused by a conformational change in CETP alternating between opened and closed states [Citation99]. Another AA MD simulation revealed a different CETP-HDL interaction mechanism with a detailed picture of the interactions between the protein and the components of the HDL [Citation100]. The study reported an upright penetration of CETP into the HDL particle surface, the migration of CE from the HDL core to the CETP opening, and transfer of CE from the opening to the binding pocket of CETP [Citation100]. Both above-mentioned CETP AA simulations used GROMOS53A6 UA force field [Citation76] for the protein, and Berger parameters [Citation80] for lipids. AA MD simulations with the CHARMM force field have also been used to investigate the CE transfer pathway through CETP. The study revealed that the hydrophobic tunnel inside CETP is sufficient to allow a CE molecule to completely transfer through the entire tunnel. The predicted rate and transfer time through the tunnel were comparable with those obtained through physiological measurements [Citation101].
The α-tocopherol transfer protein (α-TTP) is a liver cytosolic transport protein that facilitates the transport of α-tocopherol to liver-secreted plasma lipoproteins and plays an important role in the efficient recycling of plasma vitamin E [Citation102] (see ). A combination of AA and CG MD study using the AMBER FF99SB with LIPID11 FF and MARTINI CG FF investigated the interaction and the binding of α-TTP to phosphatidylinositol phosphate lipids (PIPs), when the protein was embedded in a lipid bilayer mimicking the plasma membrane composition: DOPC and DOPE lipids plus either a PI(4,5)P2 or a PI(3,4)P2 molecule [Citation103]. The simulations revealed that enrichment of membranes with PIP2 molecules facilitates the anchoring of α-TTP to the membrane surface [Citation103]. The results showed that protein binding occurs by direct interaction of the negatively charged PIP2 headgroup with the positively charged residues present at the protein near the opening of the ligand-binding cavity. Detailed AA simulations also revealed additional contacts between the protein and membrane. An example of such contacts was the formation of H-bonds between protein and the polar heads of DOPC lipids. These H-bonds were formed after protein anchoring to PIP2 and could not be observed in CG simulations [Citation103].
Phosphatidylinositol-transfer proteins (PITPs) are LTPs that can catalyze the inter-membrane transfer of PI and PC in vitro [Citation104]. AA MD simulations using OPLS force field [Citation105] have also been employed to study the binding of the mammalian StART-like PI/PC transfer protein PITPα to membrane bilayers [Citation106]. The study revealed that association of PITPα with the membrane is spontaneous and captured the partial extraction of PC from bilayer into the PITPα hydrophobic pocket. Steered MD and umbrella sampling simulations were also employed to study phospholipid loading and unloading, respectively, by PITPα. This work also revealed that the presence of PITPα on the bilayer significantly reduces the free energy of desorption of PI and PC, implying the remarkable role of PITPα in facilitating lipid loading and unloading processes [Citation106].
Fatty acid-binding proteins (FABPs) are another class of LTPs. They can transfer free fatty acids and other detergent-like compounds between cellular compartments. An UA MD simulation using the GROMOS96 force field with the G53A6 parameter set was used to investigate the binding and orientation of the fatty acid-binding protein ReP1-NCXSQ to anionic membrane in low and high ionic strength using Na+ and NaCl, respectively, for neutralizing the systems [Citation25]. The results revealed the electrostatic nature of the binding of the protein to the membrane. The authors performed several simulations with different initial configurations (orientations and distances) of protein with respect to the membrane. The study showed that in anionic membranes the protein electric macrodipole alignment follows the electric field generated by the negatively charged lipid interface. They also observed that in the presence of high ionic strength the approach of the protein to the membrane was slower than in low ionic strength, indicating that the effect of the membrane electric field on the orientation was shielded by the presence of the soluble ions. They concluded that these observations show that a combination of dipole–electric field interaction and local interactions influence the binding and orientation of the ReP1-NCXSQ protein in the interface [Citation25].
2.2. Coarse grained models
CG models simplify a molecular system by decreasing the number of particles being treated explicitly during the simulations, thus drastically reducing computation time compared to all atoms models. Also, a significant advantage over simpler models, such as implicit models (Cf section 2.3) is the treatment of lipids as individual molecules, allowing for a higher level of resolution in the membrane itself. This is beneficial for simulations of lipid mixtures and realistic models of the complexity and diversity of cell membranes. There exist many CG models for simulations of lipid systems [Citation107–113]. Not all include a compatible protein model though. We focus here on three CG models that have proven useful to simulate peripheral membrane binding.
2.2.1 MARTINI
The history of the MARTINI force field starts with a model that was meant for the simulations of lipids [Citation114,Citation115]. This model was parameterized to reproduce thermodynamics properties such as oil/water partition coefficients. This top-down approach is more similar in its spirit to the implicit membrane models than to the atomistic force fields, usually parameterized to reproduce structural data. The lipid model was later extended to proteins [Citation116] and has since then been continuously expanding with new capability to become what is currently the most used CG force field for simulations of biomolecules. MARTINI uses on average one bead to represents 4 heavy atoms and their associated hydrogens (4:1 mapping). Smaller beads are also available (3:1. and 2:1 mapping) for particular cases. The beads differ by their polarity or hydrophilicity. There are eighteen types of beads in MARTINI 2.2 [Citation117], and they are sorted in four different groups: Q (charged), P (polar), N (intermediate) and C (apolar). MARTINI 2.2P is a variant with polarized side chains. The latest version of the force field, MARTINI 3, counts 29 bead types and 7 groups, adding groups for halo-compounds (X), divalent ions (D) and a separate group for water (W). Interactions between beads are represented by a Lennard-Jones potential and parameterized based on thermodynamics data. Non-bonded interactions are short-ranged with a cut-off at 11 Å. Local geometry is maintained through bonded terms parameterized on atomistic simulation data. In addition, elastic network models should be used to maintain the packing of large biomolecules such as protein or DNA [Citation118]. MARTINI-Dry is a version with an implicit solvation model [Citation119].
With ca. 4 times less particles than an atomistic simulation and shorter-range non-bonded potentials, there are fewer interactions to compute leading to the high computational efficiency of MARTINI. Time-steps for simulation can be of 20–30 fs instead of 1–4 fs for an atomistic simulation. The coarse graining implies a number of limitations one should be aware of, including the non-directionality of H-bonds, missing conformational entropy (but reduced enthalpy as a compensation), and the restricted flexibility of biomolecules. Timescale of simulated events, and therefore kinetics information, should be interpreted carefully as the atomistic frictions are absent. Driving forces should also be interpreted with care, and temperature-dependence cannot be correctly reproduced. Electrostatic screening is implicit as the dielectric constant is the same whatever the polarity of the media is (εr = 15), though there is a workaround with the polarizable water model; two charges are added to the water bead [Citation120].
Although the top-down thermodynamics-based parameterization of MARTINI reminds of the parameterization of some implicit membrane models, the level of resolution of interfacial protein-lipid interactions is higher. Comparing MARTINI 2.2P CG simulations with MD using AA CHARMM36, we showed that the CG simulations successfully reproduce the IBS, depth of anchorage and orientation of seven different PMPs [Citation121]. In combination with AA simulations, MARTINI is a powerful membrane model to predict IBS, shed light on binding mechanism and map protein-lipid interactions, as illustrated for example by the study from Charlier et al.on interfacial binding of the HIV-1 matrix protein [Citation122], or the study by Koivuniemi et al. of a cholesteryl ester transfer protein [Citation99]. MARTINI has also been used for simulations of the GRP1 PH domain which, combined to umbrella sampling PMFs, were used to evaluate the free energy of binding of the PH domain to phosphatidylinositol phosphates-containing bilayers [Citation123].
2.2.2 Residue-Based Coarse Grained model
The Residue-Based Coarse Grained model (RBCG) [Citation124] is a protein CG model which can be used with the MARTINI CG lipids [Citation114,Citation115]. RBCG uses the same lipid mapping but for proteins, the RBCG graining is coarser than MARTINI models with only two beads per amino acids: one for the backbone and one for the sidechain. The protein RBCG model counts 20 types of beads (19 side chains beads and one backbone bead) sorted in one of the 4 classes used for the MARTINI models [Citation114,Citation115]. RBCG is available for simulations with the NAMD simulation package [Citation29] and a plugin in VMD [Citation125] can be used to generate a RBCG system. While the integration timestep can be chosen between 30 and 50 fs for simulations of lipid bilayers, a 25 fs time step has been used for protein-containing systems [Citation124]. To maintain the tertiary structure, a harmonic potential with a force constant of 5 kcal.mol−1.Å2 is added between the backbone beads that are not bonded otherwise. This was parametrized against a 20 ns atomistic simulation of the BAR peripheral domain [Citation126]. For simulations of BAR domains, the authors found that a dielectric constant εr = 1 resulted in the best agreement between all-atom and RBCG simulations, even though higher values of dielectric constant are used in other studies [Citation114,Citation124,Citation127].
2.2.3 Shape-Based Coarse Grained model
The Shape-Based Coarse Grained (SBCG) model is a coarser model than RBCG and aims to simulate the movements of proteins assemblies from large-scale interactions. While its first application was to viral capsids [Citation128] and bacterial flagellum [Citation129], it has also proven useful to study membrane curvature induction by the BAR domains [Citation126,Citation130].
The graining level can be adjusted and various mapping have been used so far: from 150 atoms per beads [Citation126,Citation128,Citation130] and up to 500 atoms per beads [Citation129]. To faithfully model the shape of the protein, the Topology Representing Network is used to position the beads [Citation131–133]. The atom-beads mapping is then based on a Voronoi diagram, and the charge and mass of the beads are equal to the total charge and mass of the Voronoi cell. The topology of the system during simulations is maintained by a harmonic distance restraint between beads. Obviously, this type of coarse graining prevents conformational changes and protein refolding [Citation128].
Conversely to the MARTINI or RBCG models, the lipid graining is not done for individual lipids. Rather, a SBCG lipid represents several lipids and consists of two beads (one for the heads and one for the tails) connected by a harmonic bond. For DOPC, one SBCG lipid represents on average 2.2 lipid molecules. The solvent is treated implicitly with a Langevin term to account for water viscosity, but there are beads for Na+ and Cl− counterions. While a dielectric constant of 80 was used for simulations of viral capsid systems [Citation128] and flagellum [Citation129], εr was set to 1 for simulations of a peripheral BAR domain [Citation126,Citation130] for the same reason as those invoked for RBCG.
An integration time step of 100 fs can be used [Citation126,Citation130] and SBCG topology systems can be generated using VMD [Citation125].
2.2.4 Coarse grained modelling of sensing and induction of membrane curvature by BAR domains
Figure 6. Bar domains structures (N-BAR:1ZWW [Citation134], F-BAR:2V0O [Citation135], I-BAR:1Y20 [Citation136]) coloured by subunit. Rendered with 3DProteinImagning [Citation12]
![Figure 6. Bar domains structures (N-BAR:1ZWW [Citation134], F-BAR:2V0O [Citation135], I-BAR:1Y20 [Citation136]) coloured by subunit. Rendered with 3DProteinImagning [Citation12]](/cms/asset/1bcfb50d-4fc5-4167-b18a-142bcc18b99e/tapx_a_1932589_f0006_oc.jpg)
Thanks to their computational efficiency and the fact that they account for individual lipids, CG force fields have been the method of choice to model membrane remodelling events. BIN/amphiphysin/Rvs (BAR) domains are long, mostly helical dimeric PMPs that can vary in length, curvature, and membrane affinity depending on the BAR classification [Citation137]. N-BAR has the highest positive curvature and can form tubules that are 20 to 60 nm wide [Citation138,Citation139]. F-BAR has the lowest positive curvature and forms wider tubules (60 to 100 nm) [Citation138], and I-BAR has a negative curvature generating membrane protrusions [Citation140] (). A given BAR domain protein can either act on the membrane shape or sense the curvature of the membrane depending mainly on two parameters: local protein concentration and membrane curvature [Citation137,Citation141,Citation142].
In 2008, Arkhipov et al. studied the membrane sculpting by an N-BAR domain using methods covering four different levels of resolution [Citation126]: AA simulations with a single BAR domain protein, RBCG simulations with 6 BAR domains to explore the local and global curvature of the membrane, longer SBCG simulations to model the global membrane curvature with 6 BAR domains, and a custom continuum elastic membrane model. The authors evaluated the effect of two different arrangements of 6 N-BAR domains (aligned or staggered [Citation139]) over an initially planar membrane. The use of RBCG reduced the number of particles to be simulated down to ~200,000, and to
~4 000 particles with SBCG instead of several million atoms with an atomistic model, thus allowing extremely long simulations (up to 5 µs in SBCG). While the staggered arrangement in RBCG led to a global membrane curvature (curvature radius ~400 Å) comparable to experimentally observed values [Citation143], the aligned arrangement led to the highest local membrane curvature (~150 Å) under each BAR-domains. Interestingly, the SBCG simulations also showed the formation of a global curvature for the aligned system after 1 µs but with a lower curvature radius (up to 1000 Å).
In 2013, the same team reported a spectacular investigation of the membrane tubulation process by the F-BAR domain [Citation144] using more than 30 SBCG simulations of 3 µs each to find the F-BAR lattice yielding the highest membrane curvature. Lattices with lower densities produced smaller membrane curvature and denser lattices achieve higher curvature. However, at very high density (over 10 dimers per 1000 nm2), the F-BARs dimers hindered each other access to the membrane yielding a lower curvature.
Using the lattice that achieved the highest curvature radius as a starting point, they increased the number of F-BAR domains to 68 and they performed two 350 µs SBCG simulations with a large patch of lipids (∼380x17 nm). Each system is composed of 21,800 beads, in comparison, the equivalent atomistic model would contain more than 15 million atoms. In both simulations, they could observe that the curvature of the membrane increased from the sides towards the centre to finally form a tubular structure with a radius of 60–90 nm.
For very large systems like the ones described above, the coarseness level of the SBCG model allows simulations at time-scales that cannot be reached with ‘finer’ CG force fields, even with the most recent HPC technology or GPU scaling [Citation145–149]. In 2019, Iqbal Mahmood et al. reported simulations of the F-BAR domain with the MARTINI model reaching 2 µs for a 14 F-BAR-membrane system and 10 µs for a system with 6 F-BAR plus membrane [Citation150] to study their capacity to curve the membrane or sense its curvature. The advantage of using a finer-grained CG was the need to take into account the flexibility of the F-BAR domains, which the SBCG does not account for. Since the SBCG method does not allow intrinsic conformational change within the F-BAR domain [Citation144], this recent study emphasized the flexibility of the F-BAR domain to match a variety of membrane curvature. They could show that F-BAR dimers assemble to bend the membrane collectively, but most interestingly, their simulation revealed the F-BAR curvature sensing property. It appears that F-BAR prefers a curvature that is consistent with the experimental curvature of striated tubes (53 ± 18 nm) and pearling vesicles (35 ± 5 nm) [Citation151].
2.3. Implicit models
The first membrane implicit models were developed early [Citation43,Citation152–154] to model the stability of transmembrane segments or the complexity of the water–lipid interface when computational resources did not allow atomistic simulations of lipid bilayers. Some implicit membrane models were typically building on implicit solvent models [Citation155–157].
Implicit models typically approximate membranes by a hydrophobic slab surrounded by a polar environment while the protein remains described by an atomistic model. Protein–lipid interactions are modelled by a mean-field approach. Because of their computational efficiency implicit membrane models are particularly useful in cases where extensive sampling is needed, such as simulating landing of proteins on membrane surfaces, IBS prediction, free energy calculations, or whenever one needs multiple simulation of a system to probe the effect of a particular parameter in the simulation.
Implicit membrane models have been extensively used for modelling the folding and/or insertion of peptides in membranes [Citation158–160] or the structure and stability of transmembrane segments [Citation161]. However, they have been used relatively less for simulations of peripheral proteins. Yet, and despite their simplicity, they have proven useful to predict interfacial-binding sites and identify hot-spots by characterizing the energetic contributions of individual amino acids to the overall free energy of transfer from water to membrane, at a fraction of the cost of atomistic or CG simulations.
Here, we distinguish between two types of implicit methods; those using continuum dielectric theory, such as the Poisson-Boltzman (PB) theory-based models and those using an empirical approach based on residue-specific transfer-free energies from water to apolar solvents, such as the Implicit Membrane Model 1 (IMM1) and present examples of their utilisation for the modelling of peripheral membrane proteins. We refer the reader to the excellent review articles by Grossfield [Citation162] and Feig [Citation163] for detailed descriptions of the underlying theories.
2.3.1 Models based on the Poisson-Boltzmann and Generalized Born theories
For a solute consisting of a set of spherical particles carrying charges (e.g. a protein) embedded in a low dielectric environment the resolution of the PB equation provides a rigorous framework to calculate the electrostatic contribution to the free energy of solvation (see e.g. [Citation164,Citation165].). Methods based on these theories have naturally been applied to evaluate the electrostatic-free energy of solvation of proteins or peptides in membranes [Citation166]. The solvation-free energy is actually addressed by solving this non-linear partial differential equation for the different states. However, the full PB equation being a non-linear differential equation, its resolution has to be done numerically and is computationally expensive. Even the linearized PB equation, which is valid for weak electrostatic potentials is still fairly computationally intensive, at least compared to other implicit models. There exist efficient tools numerically solving the PB equation for biomolecular systems, such as APBS [Citation167] or DELPHI [Citation168,Citation169], among others. Yet, because of the computational costs and the challenges linked to rigorous calculations of the derivatives, the PB theory has been applied mainly to one or a few structures and is not tractable for use in molecular simulations. Yet it can be useful when applied as postprocessing of selected trajectory frames. Note that non-polar interactions to account for van der Waals solute-solvent interactions and cavity creation (solvent-solvent) can be added using a term depending on the solvent-accessible surface of the protein. A particularly interesting application relevant for this review is the evaluation of the electrostatic-free energy of interaction between lipid bilayers and peripheral proteins [Citation20,Citation38,Citation170,Citation171]. This can be calculated as the difference between the electrostatic-free energy of the protein-membrane complex and the sum of the electrostatic-free energies of the protein and membrane infinitely far apart from each other. In this case the membrane lipids and protein are the solute – and treated with an atomistic model – while water and ions are treated by the continuum dielectric. Mulgrew-Nesbitt et al. have dissected the contributions of positively charged amino acids to the interaction of proteins with membrane surfaces, and evaluated the role of electrostatics for a large number of such proteins [Citation7], and found qualitatively good agreement with experimental data. Still, it is important to remember that this type of calculation has limitations pertaining mostly to the static character of the protein and lipid bilayers and the rigidity of the protein and bilayer. The membrane surface potential is also badly modelled by these approaches.
An attempt to simplify the complexity of the PB-based computations resulted in the development of popular Generalized-Born (GB) models. The GB models are based on the same principles, i.e. a description of the solvent or membrane as a continuum model, but use specific simplifying assumptions for the solvation-free energy to allow a closed-form approximated solution to be obtained. The GB formulation allows the use of analytical derivatives and is tractable for MD simulations, but the choice of method for the computation of the Born radii is decisive for the efficiency of the method (Cf [Citation162,Citation163]. and references therein). A rising interest in modelling of membrane proteins resulted in a number of adaptation and modification of GB methods to represent multiple dielectric environments and successfully applied to membranes [Citation172–174].
GBSW, Generalized Born with a switching function [Citation175], has been applied to describe the interactions between diacylglycerol derivatives and the C1b domain of protein kinase C δ (PKCδ), which are dependent of the interactions of PKCδ C1 with the membrane. The DAG analogues were docked in the C1 domain and the complexes were subjected to molecular simulations in the presence of a membrane modeled with GBSW to describe the ligands binding mode [Citation176].
GBSW-GCS includes a Gouy Chapman Stern term to describe anionic lipids [Citation177]. It was successfully used to predict the binding orientation of the DNA-repair protein RecA to anionic membranes, calculate binding free energies and correctly predict mutations weakening the affinity of RecA for anionic membranes [Citation178]. The orientations obtained with the GBSW-CGS membrane model were subsequently used as starting structures in atomistic simulations [Citation178,Citation179].
2.3.2 Implicit Membrane Model 1
IMM1 [Citation180] and its extension IMM1-GC [Citation181] to charged membranes are implemented in CHARMM [Citation182] and compatible with the CHARMM19 force field. IMM1 and IMM1-GC are models for simulations of proteins in zwitterionic and anionic membranes, respectively. IMM1 is based on the EEF1 (effective energy function) model for water-soluble proteins, which uses a linear distance-dependent dielectric constant [Citation155]. The ionic side-chains are neutralized and a Gaussian solvent exclusion term is added to the energy function of the CHARMM19 force field [Citation183].
IMM1-GC extends IMM1 using the Gouy-Chapman theory [Citation184] for the diffuse electrical double layer and thus allows for interactions between a charged bilayer and the amino acids of the protein to be accounted for. The model has been extended to include a transmembrane voltage potential [Citation185], a dipole potential for symmetric bilayers [Citation186], the description of pores for transmembrane proteins with internal aqueous pores [Citation159,Citation187]. The IMM1 model can also model the effect of lateral pressure and curvature stress in flat [Citation188] as well as in spherical and tubular membranes [Citation189]. Mori et al. developed an implicit micelle model called IMIC [Citation190] based on IMM1 where a superellipsoid function was used to model the micelle shape. They could show that the model faithfully reproduced structure and dynamics of transmembrane proteins obtained from AA MD simulations.
Simulations using IMM1 as membrane model have been used to predict the membrane-binding site of a human membrane-bound neutrophil protease called Proteinase 3 (PR3) [Citation191], and showed that the hydrophobic insertion of aromatic amino acids contributes significantly to the free energy of transfer of Proteinase 3 to zwitterionic bilayers [Citation40]. This was unlike its homologue the human Neutrophil Elastase (NE) that, according to simulations, inserted fewer hydrophobic amino acids. The modelling results concurred with subsequent Surface Plasmon Resonance experiments showing that the affinity of NE for POPC bilayers was lower than that of PR3 and driven mostly by electrostatics interactions. IMM1-GC was also used to identify the amino acids that are key to the protein–membrane interaction; those were verified experimentally using site-directed mutagenesis and protein expression in cell lines [Citation192].
Implicit models such as IMM1 are also very useful to generate starting structures for atomistic simulations of peripheral binders. Simulations with IMM1-GC were used to predict the orientation of Bacillus Thurigiensis phosphatidylinositol-specific phospholipase C which was used as starting structure for atomistic simulations [Citation3]. We also used IMM1 to predict the orientation and depth of anchorage of the C-terminus helices of the N-terminal acetyltransferase Naa60. We could identify the amino acids contributing the most to the free energy. The predicted orientation was used to initiate atomistic simulations while the predicted hot-spots for membrane binding were used to design mutants that were tested in vitro [Citation37]. More recently, Nepal et al.reported an innovative study of the ESCRT III subunit Snf7 where they could compare the energetics of the binding of monomers and oligomers of the subunit Snf7 to flat and negatively curved membranes. By doing this they could predict which curvature yields strongest binding [Citation193].
Implicit models like IMM1 are extremely computationally efficient and provide information about the thermodynamics of protein–membrane interactions, which can be helpful to identify interactions hotspots and design protein variants that can be further tested experimentally. That being said, and because implicit models do not explicitly describe interactions between lipids and amino acids but rather rely on a model where long-range electrostatics and the hydrophobic effect drive membrane binding, they are not the method of choice for proteins with fine-tuned lipid specificities or lipid-binding cavities.
3. Conclusion and perspectives
As stated in the introduction section, there exists ample structural data of PMPs but their membrane-bound state remains an elusive molecular assembly for structural biology techniques. It results in a lack of experimental data on the membrane-bound PMPs and leaves unanswered questions about the exact location of the IBS on the PMP structures, the amino acids key to the protein–lipid interactions (hot-spots), protein structural changes upon membrane binding, or structural changes to the membrane to name a few.
As demonstrated by the many examples referred to in section 2 of this review, MD simulations have proven extremely useful to address those questions and more. Indeed, molecular simulations have shed light on the dynamics of many PMPs and the thermodynamics of the protein–membrane interactions providing unvaluable mechanistic insights, something MD simulations are particularly good at, irrespective of the type of protein. A drawback with MD simulations though is the associated computational cost, scaling as a function of N2 where N is the number of particles simulated.
Molecular simulations of PMPs thus call for models representing membranes at different levels of resolution and many studies combine several models: implicit and atomistic, CG and atomistic, different CG levels, etc. There is no golden rule for which model(s) can be combined with which other(s) as long as the scales covered by the different models are relevant for the time and size scales of the system under investigation. In general, multiscale strategies should take full advantage of the wide range of models available.
In addition, different types of simulations and analyses may be needed. Simulations at different time-scales are used to address the variety of questions asked about PMPs – from HMMM or implicit simulations to predict the IBS and obtain a good model of the membrane-bound state, to atomistic equilibrium simulations of a few 100 ns for predicting lipid–protein interactions in the membrane-bound state, potentially followed by FEP calculations to characterize hot-spot residues. Sometimes microsecond-long simulations are needed for the association of a large PMP with a complex bilayer, and the subsequent membrane remodelling. Moreover, the sizes of the proteins or protein assemblies may vary from a few hundred amino acids to many thousands. Likewise, the dimensions and complexity of the lipid bilayer needed to have a realistic membrane model vary tremendously from one PMP to another.
In this review, we have described membrane models from those with the highest level of details – all atoms force fields with full description of the lipids and other membrane components – to the simplest ones, the implicit membrane models. We have illustrated what the different models are most adequate for using examples of the literature. It is important to note that the differences between models are not only relevant for their computational efficiency, but also for the type of information they can provide depending on which reference data they are parameterized against.
The emerging complexity of biological membranes is pointing at a more complex picture for PMP-membrane recognition mechanisms than the old model based on a balance between unspecific long-range electrostatics and hydrophobic insertion. There are large variations in the mechanisms employed by PMPs to achieve membrane binding and lipid specificity, and we are just starting to understand the subtle interplay between PMPs and membrane lipids. While the value of the low-resolution models is clear and undisputed given nowadays computational capabilities, high-resolution models are and will remain crucial to uncover the details of PMP-membrane interactions. Emerging methods that can reduce the computational costs of MD simulations that represent proteins and lipids at – at least – the atomistic level of details thus hold promises for future larger scale MD simulations of PMPs [Citation194,Citation195].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes
References
- Johnson JE, Cornell RB. Amphitropic proteins: regulation by reversible membrane interactions. Mol Membr Biol. 1999;16:217–35.
- Weber DK, Yao S, Rojko N, et al. Characterization of the lipid-binding site of equinatoxin II by NMR and molecular dynamics simulation. Biophys J. 2015;108:1987–1996. 10.1016/j.bpj.2015.03.024
- Grauffel C, Yang B, He T, et al. Cation-π interactions as lipid-specific anchors for phosphatidylinositol-specific phospholipase C. J Am Chem Soc. 2013;135:5740–5750.
- Gilbert GE, Kaufman RJ, Arena AA, et al. Four hydrophobic amino acids of the factor VIII C2 domain are constituents of both the membrane-binding and von Willebrand factor-binding motifs. J Biol Chem. 2002;277:6374–6381.
- Gamsjaeger R, Johs A, Gries A, et al. Membrane binding of β2-glycoprotein I can be described by a two-state reaction model: an atomic force microscopy and surface plasmon resonance study. Biochem J. 2005;389:665–673.
- Fuglebakk E, Reuter N. A model for hydrophobic protrusions on peripheral membrane proteins. PLoS Comput Biol. 2018;14:e1006325.
- Mulgrew-Nesbitt A, Diraviyam K, Wang J, et al. The role of electrostatics in protein-membrane interactions, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 2006;1761:812–826. 10.1016/j.bbalip.2006.07.002
- Gatta AT, Carlton JG. The ESCRT-machinery: closing holes and expanding roles, Curr. Opin. Cell Biol 2019;59:121–132.
- Simunovic M, Evergren E, Callan-Jones A, et al. Curving cells inside and out: roles of BAR domain proteins in membrane shaping and its cellular implications, Annu. Rev. Cell Dev. Biol 2019;35:111–129.
- Hurley JH. Membrane binding domains, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 2006;1761:805–811.
- Lemmon MA. Membrane recognition by phospholipid-binding domains, Nat. Rev. Mol. Cell Biol 2008;9:99–111.
- Tomasello G, Armenia I, Molla G. The Protein Imager: a full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics. 2020;36:2909–2911.
- Volwerk JJ, Griffith OH, Filthuth E, et al. Phosphatidylinositol-Specific Phospholipase C from Bacillus cereus at the Lipid-Water Interface: interfacial Binding, Catalysis, and Activation. Biochemistry. 1994;33:3464–3474.
- Suzuki KGN, Fujiwara TK, Edidin M, et al. Dynamic recruitment of phospholipase Cγ at transiently immobilized GPI-anchored receptor clusters induces IP3-Ca2+ signaling: single-molecule tracking study 2. J Cell Biol. 2007;177:731–742.
- Lacy MM, Baddeley D, Berro J. Single-molecule turnover dynamics of actin and membrane coat proteins in clathrin-mediated endocytosis. Elife. 2019;8:e52355.
- Subach FV, Patterson GH, Manley S, et al. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy, Nat. Methods. 2009;6:153–159.
- Harayama T, Riezman H. Understanding the diversity of membrane lipid composition, Nat. Rev. Mol. Cell Biol 2018;19:281–296.
- Allen KN, Entova S, Ray LC, et al. Monotopic Membrane Proteins Join the Fold, Trends Biochem. Sci 2019;44:7–20.
- Tsirigos KD, Govindarajan S, Bassot C, et al. Topology of membrane proteins — predictions, limitations and variations. Curr Opin Struct Biol. 2018;50:9–17. 10.1016/j.sbi.2017.10.003
- Nomikos M, Mulgrew-Nesbitt A, Pallavi P, et al. Binding of phosphoinositide-specific phospholipase C-ζ (PLC-ζ) to phospholipid membranes: potential role of an unstructured cluster of basic residues. J Biol Chem. 2007;282:16644–16653.
- Goh BC, Wu H, Rynkiewicz MJ, et al. Elucidation of Lipid Binding Sites on Lung Surfactant Protein A Using X-ray Crystallography, Mutagenesis, and Molecular Dynamics Simulations. Biochemistry. 2016;55:3692–3701.
- Hirano Y, Gao Y-G, Stephenson DJ, et al. Structural basis of phosphatidylcholine recognition by the c2–domain of cytosolic phospholipase a2α. Elife. 2019;8:e44760. 10.7554/eLife.44760
- Chon NL, Osterberg JR, Henderson J, et al. Membrane Docking of the Synaptotagmin 7 C2A Domain: computation Reveals Interplay between Electrostatic and Hydrophobic Contributions. Biochemistry. 2015;54:5696–5711.
- Galassi VV, Villarreal MA, Montich GG. Relevance of the protein macrodipole in the membrane-binding process. Interactions of Fatty-acid Binding Proteins with Cationic Lipid Membranes, PLoS One. 2018;13:pp. 1–15.
- Galassi VV, Villarreal MA, Posada V, et al. Interactions of the fatty acid-binding protein ReP1-NCXSQ with lipid membranes. Influence of the membrane electric field on binding and orientation, Biochim. Biophys. Acta - Biomembr 2014;1838:910–920.
- Monje-Galvan V, Klauda JB. Peripheral membrane proteins: tying the knot between experiment and computation, Biochim. Biophys. Acta - Biomembr 2016;1858:1584–1593.
- Muller MP, Jiang T, Sun C, et al. Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem Rev. 2019;119:6086–6161. 10.1021/acs.chemrev.8b00608
- Whited AM, Johs A. The interactions of peripheral membrane proteins with biological membranes, Chem. Phys. Lipids. 2015;192:51–59.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. 10.1002/jcc.20289
- Klauda JB, Venable RM, Freites JA, et al. Update of the CHARMM All-Atom Additive Force Field for Lipids: validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843.
- Best RB, Zhu X, Shim J, et al. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J Chem Theory Comput. 2012;8:3257–3273. 10.1021/ct300400x
- MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. 10.1021/jp973084f
- Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding, Chem. Phys Lett. 1999;314:141–151.
- Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys. 2004;120:11919–11929.
- Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J Comput Phys. 1977;23:187–199.
- Izrailev S, Stepaniants S, Isralewitz B, et al. Steered Molecular Dynamics, Springer, Berlin. Heidelberg. 1999:39–65. 10.1007/978-3-642-58360-5_2
- Aksnes H, Goris M, Strømland Ø, et al. Molecular determinants of the N-Terminal acetyltransferase Naa60 anchoring to the Golgi membrane. J Biol Chem. 2017;292:6821–6837. 10.1074/jbc.M116.770362
- Khan HM, He T, Fuglebakk E, et al. A Role for Weak Electrostatic Interactions in Peripheral Membrane Protein Binding. Biophys J. 2016;110:1367–1378. 10.1016/j.bpj.2016.02.020
- Klauda JB, Brooks BR, Pastor RW. Dynamical motions of lipids and a finite size effect in simulations of bilayers. J Chem Phys. 2006;125:144710.
- Schillinger AS, Grauffel C, Khan HM, et al. Two homologous neutrophil serine proteases bind to POPC vesicles with different affinities: when aromatic amino acids matter, Biochim. Biophys. Acta - Biomembr 2014;1838:3191–3202.
- Leonard AN, Wang E, Monje-Galvan V, et al. Developing and Testing of Lipid Force Fields with Applications to Modeling Cellular Membranes. Chem Rev. 2019;119:6227–6269.
- Wohlert J, Edholm O. Dynamics in atomistic simulations of phospholipid membranes: nuclear magnetic resonance relaxation rates and lateral diffusion. J Chem Phys. 2006;125:204703.
- Edholm O, Jähnig F. The structure of a membrane-spanning polypeptide studied by molecular dynamics. Biophys Chem. 1988;30:279–292.
- Lyubartsev AP, Rabinovich AL. Force Field Development for Lipid Membrane Simulations, Biochim. Biophys. Acta - Biomembr 2016;1858:2483–2497.
- Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave, Nat. Rev. Mol. Cell Biol 2008;9:112–124.
- Corradi V, Sejdiu BI, Mesa-Galloso H, et al. Emerging Diversity in Lipid-Protein Interactions. Chem Rev. 2019;119:5775–5848. 10.1021/acs.chemrev.8b00451
- Marrink SJ, Corradi V, Souza PCT, et al. Computational Modeling of Realistic Cell Membranes. Chem Rev. 2019;119:6184–6226.
- Erimban S, Daschakraborty S. Compatibility of advanced water models with a united atom model of lipid in lipid bilayer simulation. J Chem Phys. 2019;151:65104.
- Ohkubo YZ, Pogorelov TV, Arcario MJ, et al. Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys J. 2012;102:2130–2139.
- Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulation. J Comput Chem. 2004;25:1400–1415.
- Schlenkrich M, Brickmann J, MacKerell AD, et al. An Empirical Potential Energy Function for Phospholipids: criteria for Parameter Optimization and Applications. Biol. Membr 1996;31–81. DOI:https://doi.org/10.1007/978-1-4684-8580-6_2
- Feller SE, Yin D, Pastor RW, et al. Molecular dynamics simulation of unsaturated lipid bilayers at low hydration: parameterization and comparison with diffraction studies. Biophys J. 1997;73:2269–2279.
- Feller SE, MacKerell AD. An improved empirical potential energy function for molecular simulations of phospholipids. J Phys Chem B. 2000;104:7510–7515.
- Klauda JB, Brooks BR, MacKerell AD, et al. An ab initio study on the torsional surface of alkanes and its effect on molecular simulations of alkanes and a DPPC bilayer. J Phys Chem B. 2005;109:5300–5311.
- Klauda JB, Pastor RW, Brooks BR. Adjacent gauche stabilization in linear alkanes: implications for polymer models and conformational analysis. J Phys Chem B. 2005;109:15684–15686.
- Khan HM, Grauffel C, Broer R, et al. Improving the Force Field Description of Tyrosine–Choline Cation−π Interactions: QM Investigation of Phenol–N(Me)4+ Interactions. J Chem Theory Comput. 2016;12:5585–5595.
- Khan HM, MacKerell AD, Reuter N. Cation-π Interactions between Methylated Ammonium Groups and Tryptophan in the CHARMM36 Additive Force Field. J Chem Theory Comput. 2019;15:7–12.
- Petersen FNR, Jensen MØ, Nielsen CH. Interfacial Tryptophan Residues: a Role for the Cation-π Effect? Biophys J. 2005;89:3985–3996.
- Hirano Y, Gao YG, Stephenson DJ, et al. Structural basis of phosphatidylcholine recognition by the C2-domain of cytosolic phospholipase A2alpha. Elife. 2019;8:e44760.
- Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. 10.1002/jcc.20290
- Leong SW, Lim TS, Choong YS. Bioinformatics for Membrane Lipid Simulations: models, Computational Methods, and Web Server Tools. Bioinforma. - Updat. Featur. Appl 2016. DOI:https://doi.org/10.5772/62576
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general Amber force field. J Comput Chem. 2004;25:1157–1174.
- Jójárt B, Martinek TA. Performance of the general amber force field in modeling aqueous POPC membrane bilayers. J Comput Chem. 2007;28:2051–2058.
- Dickson CJ, Rosso L, Betz RM, et al. GAFFlipid: a General Amber Force Field for the accurate molecular dynamics simulation of phospholipid. Soft Matter. 2012;8:9617–9627.
- Skjevik ÅA, Madej BD, Walker RC, et al. LIPID11: a modular framework for lipid simulations using amber. J Phys Chem B. 2012;116:11124–11136.
- Dickson CJ, Madej BD, Skjevik ÅA, et al. Lipid14: the amber lipid force field. J. Chem. Theory Comput. 2014;10:865–879.
- Jämbeck JPM, Lyubartsev AP. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J Phys Chem B. 2012;116:3164–3179.
- Jämbeck JPM, Lyubartsev AP. An extension and further validation of an all-atomistic force field for biological membranes. J Chem Theory Comput. 2012;8:2938–2948.
- Sonne J, Jensen MØ, Hansen FY, et al. Reparameterization of all-atom dipalmitoylphosphatidylcholine lipid parameters enables simulation of fluid bilayers at zero tension. Biophys J. 2007;92:4157–4167.
- Högberg CJ, Nikitin AM, Lyubartsev AP. Modification of the CHARMM force field for DMPC lipid bilayer, J. Comput. Chem 2008;29:2359–2369.
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236.
- Siu SWI, Vácha R, Jungwirth P, et al. Biomolecular simulations of membranes: physical properties from different force fields. J Chem Phys. 2008;128:125103.
- Kulig W, Pasenkiewicz-Gierula M, Róg T. Topologies, structures and parameter files for lipid simulations in GROMACS with the OPLS-aa force field: DPPC, POPC, DOPC, PEPC, and cholesterol. Data Br 2015;5:333–336.
- Rog T, Koivuniemi A. The biophysical properties of ethanolamine plasmalogens revealed by atomistic molecular dynamics simulations. Biochim Biophys Acta - Biomembr. 2016;1858:97–103.
- Hermans J, Berendsen HJC, Van Gunsteren WF, et al. A consistent empirical potential for water–protein interactions. Biopolymers. 1984;23:1513–1518.
- Schuler LD, Daura X, Van Gunsteren WF. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J Comput Chem. 2001;22:1205–1218.
- Chandrasekhar I, Kastenholz M, Lins RD, et al. A consistent potential energy parameter set for lipids: dipalmitoylphosphatidylcholine as a benchmark of the GROMOS96 45A3 force field. Eur Biophys J. 2003;32:67–77.
- Schmid N, Eichenberger AP, Choutko A, et al. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011;40:843–856. 10.1007/s00249-011-0700-9
- Oostenbrink C, Villa A, Mark AE, et al. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656–1676.
- Berger O, Edholm O, Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72:2002–2013.
- Chiu SW, Pandit SA, Scott HL, et al. An improved united atom force field for simulation of mixed lipid bilayers. J Phys Chem B. 2009;113:2748–2763.
- Ryckaert JP, Bellemans A. Molecular dynamics of liquid n-butane near its boiling point. Chem Phys Lett. 1975;30:123–125.
- Piggot TJ, Piñeiro Á, Khalid S. Molecular dynamics simulations of phosphatidylcholine membranes: a comparative force field study. J Chem Theory Comput. 2012;8:4593–4609.
- Tieleman DP, MacCallum JL, Ash WL, et al. Membrane protein simulations with a united-atom lipid and all-atom protein model: lipid-protein interactions, side chain transfer free energies and model proteins. J Phys Condens Matter. 2006;18:S1221.
- Vermaas JV, Baylon JL, Arcario MJ, et al. Efficient Exploration of Membrane-Associated Phenomena at Atomic Resolution. J Membr Biol. 2015;248:563–582. 10.1007/s00232-015-9806-9
- Arcario MJ, Ohkubo YZ, Tajkhorshid E. Capturing spontaneous partitioning of peripheral proteins using a biphasic membrane-mimetic model. J Phys Chem B. 2011;115:7029–7037.
- Ohkubo YZ, Tajkhorshid E. Distinct Structural and Adhesive Roles of Ca2+ in Membrane Binding of Blood Coagulation Factors. Structure. 2008;16:72–81.
- Pogorelov TV, Vermaas JV, Arcario MJ, et al. Partitioning of amino acids into a model membrane: capturing the interface. J Phys Chem B. 2014;118:1481–1492.
- Baylon JL, Lenov IL, Sligar SG, et al. Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J Am Chem Soc. 2013;135:8542–8551.
- Guengerich FP. CYTOCHROME P-450 3A4: regulation and Role in Drug Metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17.
- Fisher MB, Paine MF, Strelevitz TJ, et al. The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism*†. Drug Metab Rev. 2001;33:273–297.
- Lee J, Cheng X, Swails JM, et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput. 2016;12:405–413.
- Qi Y, Cheng X, Lee J, et al. CHARMM-GUI HMMM Builder for Membrane Simulations with the Highly Mobile Membrane-Mimetic Model. Biophys J. 2015;109:2012–2022. 10.1016/j.bpj.2015.10.008
- Vidyashankara WISJTK, Iyer G. Software News and Updates Gabedit — a Graphical User Interface for Computational Chemistry Softwares. J Comput Chem. 2012;32:174–182.
- Kono N, Ohto U, Hiramatsu T, et al. Impaired α-TTP-PIPs interaction underlies familial vitamin E deficiency. Science. 2013;340:1106–1110. 10.1126/science.1233508
- Rogaski B, Klauda JB. Membrane-binding mechanism of a peripheral membrane protein through microsecond molecular dynamics simulations. J Mol Biol. 2012;423:847–861.
- Shaw DE, Deneroff MM, Dror RO, et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun ACM. 2008;51:91–97. 10.1145/1364782.1364802
- Lipp NF, Gautier R, Magdeleine M, et al. An electrostatic switching mechanism to control the lipid transfer activity of Osh6p. Nat Commun. 2019;10:3926. 10.1038/s41467-019-11780-y
- Koivuniemi A, Vuorela T, Kovanen PT, et al. Lipid exchange mechanism of the cholesteryl ester transfer protein clarified by atomistic and coarse-grained simulations. PLoS Comput Biol. 2012;8:e1002299.
- Cilpa-Karhu G, Jauhiainen M, Riekkola ML. Atomistic MD simulation reveals the mechanism by which CETP penetrates into HDL enabling lipid transfer from HDL to CETP. J Lipid Res. 2015;56:98–108.
- Lei D, Rames M, Zhang X, et al. Insights into the tunnel mechanism of cholesteryl ester transfer protein through All-atom molecular dynamics simulations. J Biol Chem. 2016;291:14034–14044.
- Arita M, Nomura K, Arai H, et al. α-Tocopherol transfer protein stimulates the secretion of α-tocopherol from a cultured liver cell line through a brefeldin A-insensitive pathway. Proc Natl Acad Sci U S A. 1997;94:12437–12441.
- Lamprakis C, Stocker A, Cascella M. Mechanisms of recognition and binding of α-TTP to the plasma membrane by multi-scale molecular dynamics simulations, Front. Mol. Biosci 2015;2:36.
- Wirtz KWA. Phospholipid transfer proteins. Annu Rev Biochem. 1991;60:73–99.
- Jorgensen WL, Tirado-Rives J. The OPLS Potential Functions for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J Am Chem Soc. 1988;110:1657–1666.
- Grabon A, Orłowski A, Tripathi A, et al. Dynamics and energetics of the mammalian phosphatidylinositol transfer protein phospholipid exchange cycle. J Biol Chem. 2017;292:14438–14455. 10.1074/jbc.M117.791467
- Orsi M, Essex JW. The ELBA force field for coarse-grain modeling of lipid membranes. PLoS One. 2011;6:1–22.
- Seo S, Shinoda W, Force SPICA. Field for Lipid Membranes: domain Formation Induced by Cholesterol. J Chem Theory Comput. 2019;15:762–774.
- Ayton GS, Lyman E, Voth GA. Hierarchical coarse-graining strategy for protein-membrane systems to access mesoscopic scales. Faraday Discuss. 2009;144:347–357.
- Shinoda W, DeVane R, Klein ML. Multi-property fitting and parameterization of a coarse grained model for aqueous surfactants. Mol. Simul 2007;33:27–36.
- Barrera EE, Machado MR, Pantano S, et al. Coarse-Grained Phospholipids to Explore Membrane-Protein Dynamics. J Chem Theory Comput. 2019;15:5674–5688.
- Darré L, Machado MR, Brandner AF, et al. SIRAH: a structurally unbiased coarse-grained force field for proteins with aqueous solvation and long-range electrostatics. J Chem Theory Comput. 2015;11:723–739.
- Wan CK, Han W, Wu YD. Parameterization of PACE force field for membrane environment and simulation of helical peptides and helix-helix association. J Chem Theory Comput. 2012;8:300–313.
- Marrink SJ, De Vries AH, Mark AE. Coarse Grained Model for Semiquantitative Lipid Simulations. J Phys Chem B. 2004;108:750–760.
- Marrink SJ, Risselada HJ, Yefimov S, et al. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–7824.
- Monticelli L, Kandasamy SK, Periole X, et al. The MARTINI coarse-grained force field: extension to proteins. J Chem Theory Comput. 2008;4:819–834.
- De Jong DH, Singh G, Bennett WFD, et al. Improved parameters for the martini coarse-grained protein force field. J Chem Theory Comput. 2013;9:687–697. 10.1021/ct300646g
- Periole X, Cavalli M, Marrink SJ, et al. Combining an elastic network with a coarse-grained molecular force field: structure, dynamics, and intermolecular recognition. J Chem Theory Comput. 2009;5:2531–2543.
- Arnarez C, Uusitalo JJ, Masman MF, et al. Dry martini, a coarse-grained force field for lipid membrane simulations with implicit solvent. J Chem Theory Comput. 2015;11:260–275. 10.1021/ct500477k
- Yesylevskyy SO, Schäfer LV, Sengupta D, et al. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput Biol. 2010;6:1–17.
- Khan HM, Souza PCT, Thallmair S, et al. Capturing Choline-Aromatics Cation-πInteractions in the MARTINI Force Field. J Chem Theory Comput. 2020;16:2550–2560. 10.1021/acs.jctc.9b01194
- Charlier L, Louet M, Chaloin L, et al. Coarse-grained simulations of the HIV-1 matrix protein anchoring: revisiting its assembly on membrane domains. Biophys J. 2014;106:577–585.
- Naughton FB, Kalli AC, Sansom MSP. Association of Peripheral Membrane Proteins with Membranes: free Energy of Binding of GRP1 PH Domain with Phosphatidylinositol Phosphate-Containing Model Bilayers. J Phys Chem Lett. 2016;7:1219–1224.
- Shih AY, Arkhipov A, Freddolino PL, et al. Coarse Grained Protein−Lipid Model with Application to Lipoprotein Particles†. J Phys Chem B. 2006;110:3674–3684.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38.
- Arkhipov A, Yin Y, Schulten K. Four-scale description of membrane sculpting by BAR domains. Biophys J. 2008;95:2806–2821.
- Shih AY, Freddolino PL, Arkhipov A, et al. Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations. J Struct Biol. 2007;157:579–592.
- Arkhipov A, Freddolino PL, Schulten K. Stability and Dynamics of Virus Capsids Described by Coarse-Grained Modeling. Structure. 2006;14:1767–1777.
- Arkhipov A, Freddolino PL, Imada K, et al. Coarse-grained molecular dynamics simulations of a rotating bacterial flagellum. Biophys J. 2006;91:4589–4597.
- Yin Y, Arkhipov A, Schulten K. Simulations of Membrane Tubulation byLattices of Amphiphysin N-BAR Domains. Structure. 2009;17:882–892.
- Doherty N, Ritter H, Martinetz T, et al. Neural Computation and Self-Organising Maps: an Introduction. J. Oper. Res. Soc 1994;45:240.
- Martinetz TM, Berkovich SG, Schulten KJ. “Neural-Gas” Network for Vector Quantization and its Application to Time-Series Prediction. IEEE Trans. Neural Networks. 1993;4:558–569.
- Martinetz T, Schulten K. Topology representing networks. Neural Networks. 1994;7:507–522.
- Weissenhorn W. Crystal structure of the endophilin-A1 BAR domain. J Mol Biol. 2005;351:653–661.
- Henne WM, Kent HM, Ford MGJ, et al. Structure and Analysis of FCHo2 F-BAR Domain: a Dimerizing and Membrane Recruitment Module that Effects Membrane Curvature. Structure. 2007;15:839–852.
- Inanobe A, Furukawa H, Gouaux E. Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron. 2005;47:71–84.
- Simunovic M, Voth GA, Callan-Jones A, et al. When Physics Takes Over: BAR Proteins and Membrane Curvature. Trends Cell Biol. 2015;25:780–792.
- Qualmann B, Koch D, Kessels MM. Let’s go bananas: revisiting the endocytic BAR code. Embo J. 2011;30:3501–3515.
- Frost A, Perera R, Roux A, et al. Structural Basis of Membrane Invagination by F-BAR Domains. Cell. 2008;132:807–817. 10.1016/j.cell.2007.12.041
- Saarikangas J, Zhao H, Pykäläinen A, et al. Molecular Mechanisms of Membrane Deformation by I-BAR Domain Proteins. Curr Biol. 2009;19:95–107. 10.1016/j.cub.2008.12.029
- Simunovic M, Voth GA. Membrane tension controls the assembly of curvature-generating proteins. Nat Commun. 2015;6:1–8.
- Sorre B, Callan-Jones A, Manzi J, et al. Nature of curvature coupling of amphiphysin with membranes depends on its bound density. Proc Natl Acad Sci U S A. 2012;109:173–178. 10.1073/pnas.1103594108
- Peter BJ, Kent HM, Mills IG, et al. BAR Domains as Sensors of Membrane Curvature: the Amphiphysin BAR Structure. Science. 2004;303:495–499.
- Yu H, Schulten K. Membrane Sculpting by F-BAR Domains Studied by Molecular Dynamics Simulations. PLoS Comput Biol. 2013;9:1002892.
- Kutzner C, Páll S, Fechner M, et al. More bang for your buck: improved use of GPU nodes for GROMACS 2018. J Comput Chem. 2019;40:2418–2431.
- Abraham MJ, Murtola T, Schulz R, et al. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25.
- Lee TS, Cerutti DS, Mermelstein D, et al. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: performance Enhancements and New Features. J Chem Inf Model. 2018;58:2043–2050.
- Stone JE, Hardy DJ, Ufimtsev IS, et al. GPU-accelerated molecular modeling coming of age. J Mol Graph Model. 2010;29:116–125.
- Phillips JC, Hardy DJ, Maia JDC, et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020;153:044130. 10.1063/5.0014475
- Mahmood MI, Noguchi H, Okazaki K. Curvature induction and sensing of the F-BAR protein Pacsin1 on lipid membranes via molecular dynamics simulations. Sci Rep. 2019;9:14557.
- Wang Q, Navarro MVAS, Peng G, et al. Molecular mechanism of membrane constriction and tubulation mediated by the F-BAR protein Pacsin/ Syndapin. Proc Natl Acad Sci U S A. 2009;106:12700–12705.
- Ducarme P, Rahman M, Brasseur R. IMPALA: a simple restraint field to simulate the biological membrane in molecular structure studies. Proteins Struct. Funct. Genet 1998;30:357–371.
- Ben-Tal N, Honig B. Helix-helix interactions in lipid bilayers. Biophys J. 1996;71:3046–3050.
- Sanders CR, Schwonek JP. An approximate model and empirical energy function for solute interactions with a water-phosphatidylcholine interface. Biophys J. 1993;65:1207–1218.
- Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins Struct. Funct. Genet 1999;35:133–152.
- Schaefer M, Karplus M. A comprehensive analytical treatment of continuum electrostatics. J Phys Chem. 1996;100:1578–1599.
- Clark Still W, Tempczyk A, Hawley RC, et al. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J Am Chem Soc. 1990;112:6127–6129.
- Im W, Brooks CL. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc Natl Acad Sci U S A. 2005;102:6771–6776.
- Mihajlovic M, Lazaridis T. Antimicrobial peptides bind more strongly to membrane pores. Biochim Biophys Acta - Biomembr. 2010;1798:1494–1502.
- Lipkin R, Lazaridis T. Computational studies of peptide-induced membrane pore formation. Philos Trans R Soc B Biol Sci. 2017;372:20160219.
- Yuzlenko O, Lazaridis T. Membrane protein native state discrimination by implicit membrane models. J Comput Chem. 2013;34:731–738.
- Grossfield A. Chapter 5 Implicit Modeling of Membranes. Curr Top Membr. 2008;60:131–157.
- Feig M. Implicit membrane models for membrane protein simulation. Methods Mol Biol. 2008;443:181–196.
- Sharp KA, Honig B. Calculating total electrostatic energies with the nonlinear Poisson-Boltzmann equation. J Phys Chem. 1990;94:7684–7692.
- Gallagher K, Sharp K. Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys J. 1998;75:769–776.
- Ben-Tal N, Ben-Shaul A, Nicholls A, et al. Free-energy determinants of α-helix insertion into lipid bilayers. Biophys J. 1996;70:1803–1812.
- Baker NA, Sept D, Joseph S, et al. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041.
- Rocchia W, Alexov E, Honig B. Extending the Applicability of the Nonlinear Poisson−Boltzmann Equation: multiple Dielectric Constants and Multivalent Ions†. J Phys Chem B. 2001;105:6507–6514.
- Rocchia W, Sridharan S, Nicholls A, et al. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J Comput Chem. 2002;23:128–137.
- Wang J, Gambhir A, McLaughlin S, et al. Model for the Electrostatic Sequesration of PI(4,5)P 2 by Membrane-Adsorbed Basic Peptides. Biophys J. 2004;86:1969–1986.
- Murray D, McLaughlin S, Honig B. The Role of Electrostatic Interactions in the Regulation of the Membrane Association of G Protein βγ Heterodimers. J Biol Chem. 2001;276:45153–45159.
- Spassov VZ, Yan L, Szalma S. Introducing an implicit membrane in generalized born/solvent accessibility continuum solvent models. J Phys Chem B. 2002;106:8726–8738.
- Im W, Feig M, Brooks CL. An Implicit Membrane Generalized Born Theory for the Study of Structure, Stability, and Interactions of Membrane Proteins. Biophys J. 2003;85:2900–2918.
- Tanizaki S, Feig M. A generalized Born formalism for heterogeneous dielectric environments: application to the implicit modeling of biological membranes. J Chem Phys. 2005;122:124706.
- Im W, Lee MS, Brooks CL. Generalized Born Model with a Simple Smoothing Function. J Comput Chem. 2003;24:1691–1702.
- Choi Y, Pu Y, Peach ML, et al. Conformationally constrained analogues of diacylglycerol (DAG). 28. DAG-dioxolanones reveal a new additional interaction site in the C1b domain of PKCδ. J Med Chem. 2007;50:3465–3481. 10.1021/jm0702579
- Zhang L, Yethiraj A, Cui Q. Free energy calculations for the peripheral binding of proteins/peptides to an anionic membrane. 1. implicit membrane models. J Chem Theory Comput. 2014;10:2845–2859.
- Rajendram M, Zhang L, Reynolds BJ, et al. Anionic Phospholipids Stabilize RecA Filament Bundles in Escherichia coli. Mol Cell. 2015;60:374–384. 10.1016/j.molcel.2015.09.009
- Zhang L, Rajendram M, Weibel DB, et al. Ionic Hydrogen Bonds and Lipid Packing Defects Determine the Binding Orientation and Insertion Depth of RecA on Multicomponent Lipid Bilayers. J Phys Chem B. 2016;120:8424–8437.
- Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins Struct. Funct. Genet 2003;52:176–192.
- Lazaridis T. Implicit solvent simulations of peptide interactions with anionic lipid membranes. Proteins Struct. Funct. Genet 2005;58:518–527.
- Brooks BR, Brooks CL, Mackerell AD, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614.
- Neria E, Fischer S, Karplus M. Simulation of activation free energies in molecular systems. J Chem Phys. 1996;105:1902–1921.
- McLaughlin S. The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem 1989;18:113–136.
- Mottamal M, Lazaridis T. Voltage-dependent energetics of alamethicin monomers in the membrane. Biophys Chem. 2006;122:50–57.
- Zhan H, Lazaridis T. Influence of the membrane dipole potential on peptide binding to lipid bilayers. Biophys Chem. 2012;161:1–7.
- Lazaridis T. Structural determinants of transmembrane β-barrels. J Chem Theory Comput. 2005;1:716–722.
- Zhan H, Lazaridis T. Inclusion of lateral pressure/curvature stress effects in implicit membrane models. Biophys J. 2013;104:643–654.
- Nepal B, Leveritt J, Lazaridis T. Membrane Curvature Sensing by Amphipathic Helices: insights from Implicit Membrane Modeling. Biophysical Journal. 2018;114:2128–2141.
- Mori T, Sugita Y. Implicit Micelle Model for Membrane Proteins Using Superellipsoid Approximation. J Chem Theory Comput. 2020;16:711–724.
- Hajjar E, Mihajlovic M, Witko-Sarsat V, et al. Computational prediction of the binding site of proteinase 3 to the plasma membrane. Proteins Struct. Funct. Genet 2008;71:1655–1669.
- Kantari C, Millet A, Gabillet J, et al. Molecular analysis of the membrane insertion domain of proteinase 3, the Wegenerˈs autoantigen, in RBL cells: implication for its pathogenic activity. J Leukoc Biol. 2011;90:941–950.
- Nepal B, Sepehri A, Lazaridis T. Mechanisms of negative membrane curvature sensing and generation by ESCRT III subunit Snf7. Protein Sci. 2020;29:1473–1485.
- Zhang L, Han J, Wang H, et al. Deep Potential Molecular Dynamics: a Scalable Model with the Accuracy of Quantum Mechanics. Phys Rev Lett. 2018;120:143001.
- Noé F, Tkatchenko A, Müller KR, et al. Machine learning for molecular simulation. Annu Rev Phys Chem. 2020;71:361–390.