
Abstract
Salix paraflabellaris is a procumbent alpine plant endemic to Hengduan Mountain in Yunnan Province, China. The complete mitochondrial genome sequence of this species is a circular molecule of 637,893 bp in size, encoding 34 protein-coding genes, 22 tRNA genes, and 3 rRNA genes. Moreover, two large inverted repeat regions with length of 9557 bp and 3524 bp were identified in the genome. Phylogenetic analysis confirmed the membership of S. paraflabellaris in genus Salix L., as well as monophyly of both Populus and Salix.
Salix paraflabellaris S.D. Chao is a Salix L. (Salicaceae) species endemic to Yunnan Province of China. It is an alpine plant distributed in alpine scree slope habitat with elevations around 3500–4500 m in the Hengduan Mountain (Southeast of Qinghai-Tibetan Plateau). Salix paraflabellaris is distinct in morphology compared with other Salix species: it is a procumbent plant with stems and branches buried in soil and usually ca. 3 cm in height (Fang et al. Citation1999).
In this study, we sequenced and assembled the complete mitochondrial genome of S. paraflabellaris. The leaf sample was collected from an individual of this species from Shangri-La County, Yunnan Province, China (N 28.151, E 99.903). The voucher specimen was deposit in the Herbarium of Kunming Institute of Botany (accession number: CJH1709). Genomic DNA was isolated using a modified CTAB method (Porebski et al. Citation1997) and then fragmented and used to construct short-insert libraries (300 bp) following the manufacturer’s protocol (Illumina Inc., USA), and then paired-end sequenced (150 bp) on an Illumina Hiseq X Ten sequencer. A total of ca. 31.2 million reads were generated. Assembly was performed using NOVOPlasty v2.7.2 (Dierckxsens et al. Citation2017) with a k-mer of 39.
The mitochondrial genome of S. paraflabellaris was assembled as a closed-circular molecule of 637,893 bp in length (GenBank accession No.: MK575518) and annotated by GeSeq (Tillich et al. Citation2017). The genome contains 59 annotated genes, including 34 protein-coding genes (CDS), 22 tRNA genes, and 3 rRNA genes. All five categories of mitochondrial protein-coding genes that are typically conserved among plants (Chen et al. Citation2017) are present in our assembled genome. Interestingly, we identified two large repeats with a lengt of more than 500 bp using Repeat Finder implemented in Geneious v9.1.4 (Kearse et al. Citation2012). Both repeats have a frequency of 2 and are presented as inverted repeats with 9557 bp and 3524 bp in length, respectively, and no genes or protein-coding sequences harbored in these two repeats. This kind of large repeats was not found in the other four species of Salicaceae with complete mitochondrial genome available in GenBank.
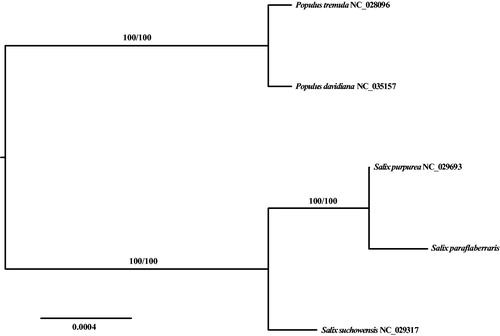
To confirm the phylogenetic position of S. paraflabellaris, we downloaded four Salicaceae species with mitochondrial complete genome from GenBank, and 31 mitochondrial CDS shared by all these five species were aligned using MUSCLE (Edgar Citation2004) and maximum likelihood (ML) tree was constructed using IQ_TREE v1.6.10 (Nguyen et al. Citation2015), best-fitted model according to Bayesian information criterion is k3Pu + F using ModelFinder (Kalyaanamoorthy et al. Citation2017), branch supports were tested by ultrafast bootstrap (UFBoot) (Hoang et al. Citation2018) and SH-like approximate likelihood ratio test (SH-aLRT) (Guindon et al. Citation2010) with 10,000 replicates. The ML tree showed that both Populus and Salix are robust monophyletic clade and sister to each other. This relationship is consistent with former studies (Huang et al. Citation2017). Salix paraflabellaris is nested in the Salix clade and sister to S. purpurea ().
Figure 1. ML phylogenetic tree of S. paraflabellaris and four Salicaceae species based on 31 mitochondrial CDS (atp-1,4, 6, 8, 9; ccm-B, C, Fc, Fn; cob; cox-1, 2, 3; matR; mttB; nad-1, 2, 3, 4, 4L, 5, 6, 7, 9; rpl-2, 10; rps-3, 4, 7, 12; sdh4) shared by the five Salicaceae species, branch supports values were reported as SH-aLRT/UFBoot.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Chen ZW, Zhao N, Li SS, Grover CE, Nie HS, Wendel JF, Hua JP. 2017. Plant mitochondrial genome evolution and cytoplasmic male sterility. Crit Rev Plant Sci. 36:55–69.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797.
- Fang Z, Zhao S, Skvortsov AK. 1999. Salicaceae. Flora of China. 4:139–274.
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59:307–321.
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35:518–522.
- Huang Y, Wang J, Yang YP, Fan CZ, Chen JH. 2017. Phylogenomic analysis and dynamic evolution of chloroplast genomes in Salicaceae. Frontiers in Plant Science. 8:1050. doi:10.3389/fpls.2017.01050
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14:587.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Porebski S, Bailey LG, Baum BR. 1997. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep. 15:8–15.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45:W6–W11.