
Abstract
In this study, we presented the complete mitochondrial genome of Pseudozyma sp. JCC207. It has a total length of 48,917 bp, and the base composition of the mitogenome is as follows: A (34.1%), T (35.4%), C (15.4%), and G (15.1%). The mitogenome contains 15 protein-coding genes, 2 ribosomal RNA (rRNA) and 20 transfer RNA (tRNA) genes. Except nad6, all protein-coding genes were located on the same strand. The genome structure is unique compared to other Ustilaginomycetes mitogenomes. The taxonomic status of the Pseudozyma sp. JCC207 mitogenome exhibits a closest relationship with Sporisorium scitamineum (CP010939), but varied in the structure of mitochondrial genome.
The specimen (Pseudozyma sp. JCC207) was first isolated from the seawater near Guam, USA in October 2004 (Mi-Hee et al. Citation2008) and was stored in Sichuan Academy of Agricultural Sciences (No. SAAS1). The total genomic DNA of obtained mycelia was extracted using Fungal DNA Kit D3390-00 (Omega Bio-Tek, Norcross, GA) according to the manufacturer’s instructions and was stored in the sequencing company (HuiTong Tech, Shenzhen, China). Purified DNA was fragmented and used to construct the sequencing libraries following the instructions of NEBNext® Ultra™ II DNA Library Prep Kit (NEB, BJ, and CN). Whole genomic sequencing was performed by the Illumina HiSeq 2500 Sequencing Platform (Illumina, San Diego, CA). Adapters and low-quality reads were removed using the NGS QC Toolkit (Patel and Jain Citation2015). Then assembly as implemented by SPAdes version 3.11.0 (Bankevich et al. Citation2012). Gaps among contigs were filled by using MITObim version 1.9 (Hahn et al. Citation2013).
The determined genome was annotated using the MFannot tool (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl), combined with manual corrections. All tRNAs were confirmed using the tRNAscan-SE search server (Lowe and Eddy Citation1997). This complete mitochondrial genome sequence together with gene annotations were submitted to GenBank under the accession numbers of MK714018.
The complete mitochondrial genome has a total length of 48,917 bp, and the base composition of the mitogenome is as follows: A (34.1%), T (35.4%), C (15.4%), and G (15.1%). The mitogenome contains 15 protein-coding genes, 2 ribosomal RNA (rRNA), and 20 transfer RNA (tRNA) genes. Except nad6, all protein-coding genes were located on the same strand.
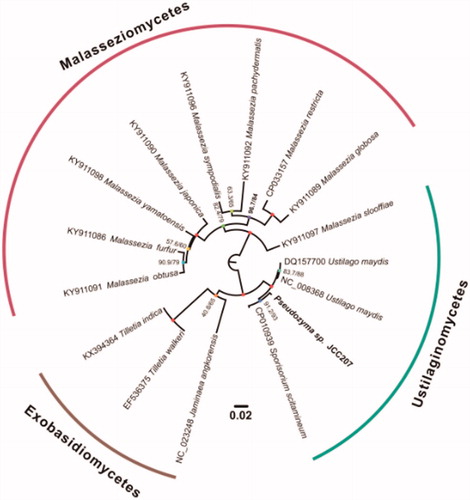
To validate the phylogenetic position of Pseudozyma sp. JCC207, the genome-wide alignment of 16 Ustilaginomycotina mitogenomes was constructed by HomBlocks (Bi et al. Citation2018). The whole genome alignment was analysed by IQ-TREE version 1.6.6 (Nguyen et al. Citation2015) under the TVM + F+R2 model. The tree topology was verified under both 1000 bootstrap and 1000 replicates of SH-aLRT test. As shown in , the phylogenetic positions of these 16 mt genomes were successfully resolved with high bootstrap and SH-aLRT supports except with two nodes. The taxonomic status of the Pseudozyma sp. JCC207 mitogenome exhibits a close relationship with Sporisorium scitamineum (CP010939), but varied in the structure of mitochondrial genome.
Figure 1. Phylogenetic tree yielded by IQ-TREE of 16 Ustilaginomycotina mitogenomes. Consensus tree is shown with support indicated by numbers at branches, representing percentages of SH-aLRT test and bootstraps.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110:18–22.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129–e129.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- Mi-Hee Chang, Hyeon-Jin Kim, Kwang-Yeop Jahng, Seong-Chool Hong. 2008. The isolation and characterization of Pseudozyma sp. JCC 207, a novel producer of squalene. Applied microbiology and biotechnology 78:963.
- Nguyen LT, Schmidt HA, Haeseler A, von Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Patel RK, Jain M. 2015. NGS QC toolkit: a platform for quality control of next-generation sequencing data. PloS one 7.2: e30619.