
Abstract
Mugil cephalus Linnaeus, 1758 is a teleost fish widely distributed in coastal waters that plays an important role in commercial fisheries. In the present study, the complete mitogenome of M. cephalus from the Yellow Sea, China, was sequenced using Illumina Novaseq sequencing. The mitogenome of the M. cephalus was 16,744 bases in length (GenBank accession No. ON262567) including 2 rRNA genes, 22 tRNA genes, 13 protein-coding genes and a D-loop control region. The overall base composition of the genome was 28.2% A, 29.5% C, 26.9% T, and 15.4% G. The analysis of genetic similarity and phylogenetic relationship of M. cephalus from different geographic regions of the world indicated that the species from the Yellow Sea was most similar to NWP1 which is one of the three cryptic species of M. cephalus in Northwestern Pacific.
1. Introduction
Flathead mullet Mugil cephalus Linnaeus, 1758 (Mugiliformes: Mugilidae) is an ecologically important species with a wide distribution, and it is also the main economic income for some regions of the world (Durand et al. Citation2012). Recent studies suggest that there might be three cryptic species (NWP1, NWP2, and NWP3) of M. cephalus in the Northwestern Pacific (Shen et al. Citation2011; Durand et al. Citation2012; Whitfield et al. Citation2012; Shen et al. Citation2016). However, currently, only four complete mitogenomes of M. cephalus from limited regions have been reported: NWP1 (GenBank: AP002930; Miya et al. Citation2001), NWP2 (GenBank: KM368340), NWP3 (GenBank: KP861225; Shen et al. Citation2011) and Eastern Australia (hereafter called from EA; GenBank: KP018403; Shen et al. Citation2016). Despite the ecological and economical importance of M. cephalus (Harrison and Howes Citation1991), the genetic resources and evolutionary relationships of the species from different geographic regions remain largely unknown. In the present study, we provided a new mitogenome of M. cephalus from the Yellow Sea, China (hereafter called from YS) and conducted a genetic comparison and phylogentic analysis of the species from different geographic regions. The study will provide a valuable genetic resource for future evolution study of M. cephalus.
2. Materials and methods
2.1. Sample collection and DNA extraction
A tail muscle sample was taken from a dead mature individual of M. cephalus () collected from the coastal waters of Qingdao (120°22′44″E, 36°18′0″N) in 2019. The specimen was deposited at the collection center of Chongqing Key Laboratory of Animal Biology (https://www.cqnu.edu.cn/, Haitao Li, [email protected]) under voucher number MG-qd-201908. Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Germany) following the manufacturer’s protocol.
Figure 1. Species image of Mugil cephalus. The dead mature individual was collected by Lei Gao from the Coast of Qingdao (120°22′44″ E, 36°18′0″ N) in 2019 which body’s length was about 20 cm, fully possessed the characteristics of M. cephalus. This photo was taken in Qingdao in 2019, by Lei Gao with the author’s approval for use.

2.2. Genome sequencing, assembly, and annotation
The complete mitogenome of M. cephalus was sequenced using the Illumina Novaseq 6000 platform as paired-end 2 × 150 base reads. A total of 76.8 G read bases of filtered data in 255,983,845 reads were assembled using Getorganelle (v1.7.7.0; Jin et al. Citation2020). The mitochondrial genome was annotated using MitoFish (http://mitofish.aori.u-tokyo.ac.jp/; Wataru et al. Citation2013) and then further checked using an existing annotated mitogenome of M. cephalus (Shen et al. Citation2016). The mitochondrial genome map of M. cephalus was constructed using MitoFish. The mitogenome read coverage depth map is shown in Supplementary Figure S1. The raw data, final genome assembly sequence, and annotated information were deposited in the National Center for Biotechnology Information (NCBI), with the accession number SRR21493539.
2.3. Genetic similarity analysis
The genetic similarities were calculated from complete mitochondrial DNA sequences and entire amino acid sequences (which were estimated from the nucleotide sequences) among M. cephalus from five localities (EA, NWP1, NWP2, NWP3, and YS) using the EMBOSS Needle.suite with the algorithm of Needleman–Wunsch (https://www.ebi.ac.uk/Tools/psa/; Rice et al. Citation2000).
2.4. Phylogenetic analysis
The dataset for phylogenetic tree reconstruction was composed of 13 of the concatenated protein-coding genes of M. cephalus and 23 other related species. Multiple alignments of the sequences of the dataset were conducted by Clustal X (Thompson et al. Citation1997) under the setting of the default parameters. MrBayes 3.1.2 (Ronquist et al. Citation2012) was employed to infer the phylogenetic tree, and the best fitting model (GTR + I + G) was selected by Modeltest 3.7 under the Akaike information criterion. Four independent Markov chain Monte Carlo ran simultaneously for 10 million generations, sampling one tree per 200 generations, and the first 25% of the samples were discarded as the burn-in. Chain convergence and parameter mixing were checked by Tracer v1.3 (Rambaut and Drummond Citation2005) based on the average standard deviation of split frequencies (0.004733).
3. Results
3.1. Characteristics of the mitogenomes
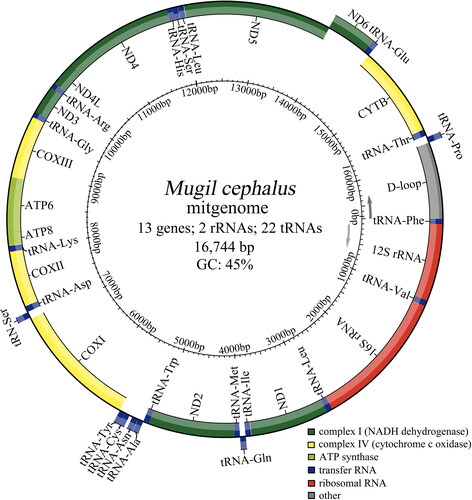
The complete mitogenome of M. cephalus was 16,744 bp (GenBank accession No. ON262567) with a base composition of 28.2% A, 29.5% C, 26.9% T, and 15.4% G. The genome possessed 13 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes, and one control region (). Among them, ND6 and eight tRNA genes (tRNA-Gln, tRNA-Ala, tRNA-Asn, tRNA-Cys, tRNA-Tyr, tRNA-Ser, tRNA-Glu, and tRNA-Pro) were encoded on light strand, and the remaining genes were encoded on heavy strand (). The gene numbers and the order of the mitogenome were consistent with other vertebrates (Yang et al. Citation2013; Sun and Xu Citation2018; Zhang et al. Citation2019). All protein-coding genes used ATG as a start codon except for COI which started with GTG. There were three stop codon types for genes: TAA type for ND1, COI, ATPase8, ATPase6, ND4L, and ND5; TAG type for ND6 and, T – type for ND2, COII, COIII, ND3, ND4, and CYTB.
Figure 2. The circular-mapping mitochondrial genome of Mugil cephalus ON262567 prepared using the MitoFish web server. Genes inside the circle are transcribed clockwise, whereas those outside are transcribed counterclockwise.
3.2. Similarity among five mitogenomes
The similarities of the complete mitochondrial nucleotide sequences and amino acid sequences of M. cephalus from five localities were 94.0%–99.4% and 98.1%–99.9%, respectively. The highest similarities (99.4% for nucleotide and 99.9% for amino acid) of M. cephalus were found between YS and NWP1 ().
Table 1. Sequence similarities of mitochondrial DNA among five localities of Mugil cephalus.
3.3. Phylogenetic analysis
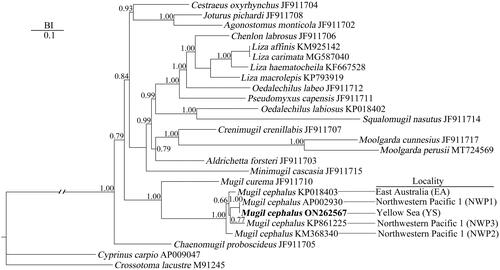
Phylogenetic analysis showed that all the M. cephalus from different localities clustered together and were sisters to M. curema (). Within the clade of M. cephalus, the YS and NWP1 were formed a clade sister to NWP3, and then to EA, and then to NWP2, successively.
Figure 3. Bayesian inference (BI) phylogenetic tree reconstruction of Mugil cephalus in Mugilidae based on the 13 concatenated nucleotide sequences of protein-coding genes, utilizing GTR + I + G as best fitting model and after running for 10 million generations. Cyprinus carpio and crossostoma lacustre were set as outgroups. The bold black font represents the species in the present study. The numbers beside the nodes are Bayesian posterior probabilities. GenBank accession numbers are beside taxon names. Behind the dotted lines are the collection sites.
4. Discussion
In the present study, we sequenced the complete mitogenome of M. cephalus from YS and conducted comparative and evolutionary analyses of the complete mitogenome with four mitogenomes of M. cephalus from other localities (NWP1, NWP2, NWP3, and EA). Mitogenome comparison and phylogenetic analysis showed that YS was most similar to NWP1. The high similarities (higher than 99%) of DNA barcode (COI) and whole sequence of mitogenome between YS and NWP1 indicated the conspecific relationship of them (). The sequence similarity analysis indicated that there are four distinct lineages: NWP1 (YS), NWP2, NWP3, and EA. As the sampling locality of YS also belongs to Northwestern Pacific, the result is consistent with the previous hypothesis that three cryptic species of M. cephalus in Northwestern Pacific (Shen et al. Citation2011; Durand et al. Citation2012; Whitfield et al. Citation2012). However, since there are still scarce of complete mitogenomes in Northwestern Pacific, broader sampling regions are needed to further test this hypothesis.
Ethical approval
The ethical approval for this study was obtained from the Animal Care and Use Committee of Chongqing Key Laboratory of Animal Biology, with the approval number CKLAB2019-009. The study was conducted in strict adherence to the ethical guidelines and regulations set forth by the committee. The authors ensured the welfare and ethical treatment of the animals involved throughout the research process.
Author contributions
Conceived and designed the study: Chengzhong Yang and Yuanjun Zhao. Wrote the manuscript: Haitao Li. Completed the experiment: Haitao Li and Lei Gao. Revised the manuscript: Chengzhong Yang, Yuanjun Zhao, and Wei Chen. Analyzed the data: Haitao Li, Wei Chen, Yanmei Huang, and Lei Gao. All authors agree to be accountable for all aspects of the work.
Supplemental Material
Download JPEG Image (872.8 KB){kind=link}
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under accession no. ON262567. The associated BioProject, Biosample, and SRA numbers are PRJNA876817, SAMN30673209, and SRR21493539, respectively.
Additional information
Funding
References
- Durand JD, Shen KN, Chen WJ, Jamandre BW, Blel H, Diop K, Nirchio M, Garcia de León FJ, Whitfield AK, Chang CW, et al. 2012. Systematics of the grey mullets (Teleostei: Mugiliformes: Mugilidae): molecular phylogenetic evidence challenges two centuries of morphology-based taxonomy. Mol Phylogenet Evol. 64(1):73–92. doi:10.1016/j.ympev.2012.03.006.
- Harrison IJ, Howes GJ. 1991. The pharyngobranchial organ of mugilid fishes; its structure, variability, ontogeny, possible function and taxonomic utility. Bull Brit Mus Nat Hist Zool. 57:111–132.
- Jin JJ, Yu WB, Yang JB, Song Y, Claude W, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. doi:10.1186/s13059-020-02154-5.
- Miya M, Kawaguchi A, Nishida M. 2001. Mitogenomic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol Biol Evol. 18(11):1993–2009. doi:10.1093/oxfordjournals.molbev.a003741.
- Rambaut A, Drummond A. 2005. Tracer-MCMC Trace Analysis Tool V.1.3. http://tree.bio.ed.ac.uk/software/tracer/.
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16(6):276–277. doi:10.1016/s0168-9525(00)02024-2.
- Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. doi:10.1093/sysbio/sys029.
- Shen KN, Chen CH, Hsiao CD, Durand JD. 2016. Next-generation sequencing yields the complete mitochondrial genome of the flathead mullet, Mugil cephalus cryptic species in East Australia (Teleostei: Mugilidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(5):3218–3219. doi:10.3109/19401736.2015.1007354.
- Shen KN, Jamandre BW, Hsu CC, Tzeng WN, Durand JD. 2011. Plio-Pleistocene sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus. BMC Evol Biol. 11(1):83. doi:10.1186/1471-2148-11-83.
- Sun YN, Xu TJ. 2018. Complete mitochondrial genome of Caranx equula (Perciformes, Carangidae): genome characterization and phylogenetic analysis. Mitochondrial DNA B Resour. 3(2):786–787. doi:10.1080/23802359.2018.1491333.
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The Clustal X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25(24):4876–4882.
- Wataru I, Tsukasa F, Ryota I, Koichiro Y, Yasunobu M, Takashi PS, Tetsuya S, Kohji M, Hirohiko T, Masaki M, et al. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Phylogenet Evol. 30:2531–2540.
- Whitfield AK, Panfili J, Durand JD. 2012. A global review of the cosmopolitan flathead mullet Mugil cephalus Linnaeus, 1758 (Teleostei: Mugilidae), with emphasis on the biology, genetics, ecology and fisheries aspects of this apparent species complex. Rev Fish Biol Fisheries. 22(3):641–681. doi:10.1007/s11160-012-9263-9.
- Yang CZ, Xiang C, Qi W, Xia S, Tu F, Zhang X, Moermond T, Yue B. 2013. Phylogenetic analyses and improved resolution of the family Bovidae based on complete mitochondrial genomes. Biochem Syst Ecol. 48:136–143. doi:10.1016/j.bse.2012.12.005.
- Zhang ZC, Cheng QQ, Ge YH. 2019. The complete mitochondrial genome of Rhynchocypris oxycephalus (Teleostei: Cyprinidae) and its phylogenetic implications. Ecol Evol. 9(13):7819–7837. doi:10.1002/ece3.5369.