
Abstract
Sepsis is a common complication after severe trauma, burns, infections, major surgery and is frequently followed by septic shock, multiple organ dysfunction syndrome, and death. Cardiac dysfunction is a non-negligible component of multiple organ dysfunction in sepsis. However, the specific mechanism of septic cardiomyopathy remains unclear. The results showed that in septic cardiomyopathy, there was damage to cardiomyocytes, while cell death was rare. A growing body of research has shown that mitochondria play an essential role in organ damage in sepsis. The severity of mitochondrial dysfunction is closely related to the prognosis of sepsis and cardiac dysfunction. To date, the mechanisms involved in mitochondrial dysfunction in septic cardiomyopathy include mitochondrial oxidative stress, Ca2+ overload, inflammatory pathways, energy deficiency, and mitochondrial dynamics. It is evident that damaged mitochondria in cardiomyocytes can be fatal to the heart. However, the exact molecular mechanism and its potential significance are still not fully understood. Here, this review intends to draw conclusions about the mechanisms associated with mitochondrial dysfunction in septic cardiomyopathy and to delve into the mechanisms involved in order to find new insights into the study of septic cardiomyopathy.
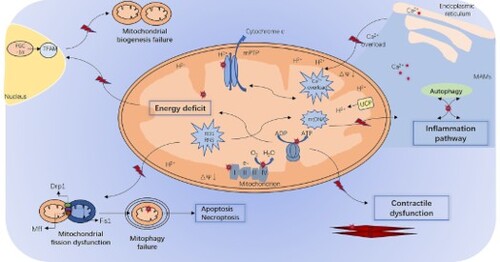
GRAPHICAL ABSTRACT
1. Septic cardiomyopathy
In 2016, the Journal of American Medical Association published a new definition of sepsis, referred to as SEPSIS-3 (Singer et al. Citation2016). Sepsis has been redefined as life-threatening organ dysfunction caused by inappropriate and excessive host response to infection (Singer et al. Citation2016). Therefore, this definition requires infection as well as acute organ dysfunction (Horak et al. Citation2019). Septic shock represents the more severe form, in which particularly severe circulatory, cellular, and metabolic abnormalities are present. The hospital mortality rate of patients with septic shock is over 40% (Beesley et al. Citation2018). 5.3 million people die from sepsis each year, which is 10% of the total cause of death in a year (Beesley et al. Citation2018). In recent years, scholars have conducted extensive research on the pathogenesis and treatment of sepsis. Various antibiotics and supportive therapies have been used, but the mortality rate is still as high as 30% (Fink and Warren Citation2014; Coopersmith et al. Citation2018). Some important reasons for the persistently high mortality rate of sepsis are the uncontrolled inflammatory response and multiple organ damage.
Cardiac dysfunction is one of the common complications of severe sepsis, also known as septic cardiomyopathy (Cimolai et al. Citation2015; Fang and Wang Citation2018). Data show that 40% to 50% of patients with sepsis have myocardial damage clinically; these patients had a longer hospital stay and higher fatality rate. Recent studies have shown that 20% of them develop severe heart failure, with a fatality rate as high as 70% (Sato et al. Citation2016; Jiang et al. Citation2019). An early animal model-based hypothesis was that septic cardiomyopathy was caused by myocardial ischemia due to decreased coronary blood flow (Bruni et al. Citation1978). Several pathophysiological mechanisms have been proposed to explain septic cardiomyopathy. One is that the cascade of sepsis leads to inflammatory and immune dysregulation, which can produce a variety of myocardial inhibitory factors, such as interleukin-16, tumor necrosis factor (TNF) and NO (nitric oxide), which then lead to cardiac dysfunction (Beesley et al. Citation2018). Moreover, currently, myocardial energy metabolism disorders, impaired cardiomyocyte calcium (Ca2+) homeostasis, and apoptosis are considered important mechanisms of myocardial dysfunction in sepsis. Cardiovascular failure can reduce overall blood circulation, which can worsen tissue hypoxia, mitochondrial dysfunction, and tissue metabolic dysfunction (Cimolai et al. Citation2015). Given the central role of circulatory dysfunction in disrupting multi-organ function, it is critical to understand the cardiac dysfunction of patients with sepsis (Beesley et al. Citation2018). However, the exact molecular mechanisms that ultimately lead to cardiac dysfunction remain unclear (Ehrman et al. Citation2018).
It has been demonstrated that cell death is rare in sepsis-induced cardiomyopathy, despite cardiomyocyte injury occurring (Takasu et al. Citation2013). It is extremely important for us to find an accurate site to repair cell function. Naturally, this turns attention to the mitochondria of cardiomyocytes. The severity of mitochondrial dysfunction is firmly associated with the prognosis of sepsis (Brealey et al. Citation2002). A growing body of research suggests that mitochondria play an important role in organ damage in sepsis by producing reactive oxygen species (ROS) and regulating cell death (Durand et al. Citation2017; Fang and Wang Citation2018).
It is clear that the mitochondrial dysfunction of myocardial cells is lethally detrimental to the heart (Pan, Wang, et al. Citation2018). This review will summarize the mechanism of mitochondrial dysfunction in septic cardiomyopathy, with a view to providing some new insights into septic cardiomyopathy.
2. Mitochondrial dysfunction in septic cardiomyopathy
The heart is the most energy-intensive organ in the body and derives substantially all of its energy from mitochondrial oxidative phosphorylation (OXPHOS) (Takasu et al. Citation2013). It is, therefore, necessary to study mitochondrial dysfunction in septic cardiomyopathy.
Myocardial depression is increasingly common and can be defined as a reversible dysfunction of the heart. Mitochondrial dysfunction can lead to sepsis-associated myocardial depression (Antonucci et al. Citation2014). The initial cause might be attributed to hypoxia (Anderson et al. Citation1946; Hotchkiss and Karl Citation1992; Lemasters et al. Citation2009). During sepsis, hypoxia caused by hypoperfusion increases free radical production due to oxygen limitation and incomplete OXPHOS (Zhang et al. Citation2018). On the other hand, the activity and expression of molecules in the antioxidant system are impaired (Zhang et al. Citation2018). After that, the production of reactive oxygen and nitrogen species increases, leading to energetic and structural failure of the cardiomyocytes (Tsolaki et al. Citation2017). Studies have shown that lipopolysaccharide (LPS) stimuli could induce NAPDH oxidase expression and production of ROS (Fink Citation2001), leading to the overproduction of RNS (reactive nitrogen species) and NO by the promotion of iNOS (inducible nitric oxide synthase) activity (Trumbeckaite et al. Citation2001; Belcher et al. Citation2002; Singer Citation2014). NO can peroxidize with ROS components to form RNS components, resulting in irreversible inhibition of electron transport chain (ETC) activity (Moncada and Erusalimsky Citation2002). In animal models and patients, iNOS inhibitors, such as melatonin, have been shown to block NO formation, thereby improving sepsis prognosis (Escames et al. Citation2006; Poggi and Dani Citation2018). This might be related to melatonin having numerous antioxidant effects (Hardeland Citation2018). Research has also shown that melatonin might protect cardiomyocytes through modulating uncoupling protein-2(UCP2), which might play a protective role against LPS by regulating autophagy and apoptosis of cardiomyocytes (Pan, Zhang, et al. Citation2018).
As mentioned above, the accumulation of mitochondrial ROS may impair ETC function and ETC complexes, which leads to insufficient intracellular ATP production (Piel et al. Citation2008). This might be the main cause of myocardial contractility decline. Mitochondria may result in further damage, including endometrial damage and mtDNA damage, thereby inducing Ca2+ reflux and cytochrome c release and subsequent apoptosis in cardiomyocytes (Lemasters et al. Citation2009). At higher ROS levels, due to the Ca2+ overload, prolonged mitochondrial permeability transition pore (mPTP) openings might release a ROS burst leading to the destruction of mitochondria, cessation of ATP synthesis, and then propagation from mitochondrion to mitochondrion (Zorov et al. Citation2014).
In conclusion, membrane instability or disruption caused by morphological changes is a promoter of apoptosis and deranged Ca2+, while incomplete OXPHOS caused by mitochondrial dysfunction can cause energy deficiency and ROS overload, which leads to cell damage (Zhang et al. Citation2018). Finally, the mitochondria undergo matrix swelling, membrane rupture and enhanced apoptosis. As for the evaluation of mitochondrial morphological changes, the rat endotoxin-induced peritonitis model established by Vanasco et al. reported abnormalities in myocardial mitochondria at 6 and 18 h after LPS injection, such as swelling, loss of cristae, cleared matrix, and membrane rupture (Vanasco et al. Citation2014). In septic conditions, mitochondrial dysfunction is typically described as above. Mitochondrial dysfunction can promote mitochondrial damage, decrease of mitochondrial ATP synthesis, and lead to myocardial cell death through mitochondrial dependent apoptosis pathway, thus failing to support heart function in septic cardiomyopathy (Cao et al. Citation2020; Jiang et al. Citation2021). Regulating specific molecules to improve mitochondrial dysfunction in septic cardiomyopathy can enhance mitochondrial autophagy and biosynthesis, maintain the stability of mitochondrial fission and fusion, reduce endoplasmic reticulum (ER) stress and inflammatory response, and ultimately protect the heart in sepsis (Zhong, Tan, et al. Citation2019; Zhang et al. Citation2020; Li et al. Citation2021; Qiao et al. Citation2021). Mitochondrial quality control (MQC) coordinates various processes (division, fusion, mitochondrial autophagy, and mitochondria-controlled cell death) to ensure cell homeostasis, which may function as molecular targets (Wang and Zhou Citation2020). Therefore, mitochondrial dysfunction is a potential mechanism of septic cardiomyopathy (Levy Citation2007; Fender et al. Citation2020). It should be noted that mitochondrial dysfunction in septic cardiomyopathy is neither a cause nor a consequence but rather a process that plays an amplifying role in the vicious cycle of the pathophysiological process of sepsis (Zhang et al. Citation2018).
3. Molecular mechanisms of mitochondrial dysfunction in septic cardiomyopathy
3.1. Mitophagy-related molecular pathways
Autophagy might play a protective role in maintaining intracellular homeostasis in cardiomyocytes (Bravo-San Pedro et al. Citation2017). Current studies have shown that the hypophosphorylation form of the IκBβ Ser313 site is beneficial to the heart in sepsis by inhibiting apoptosis and enhancing autophagy (Wang et al. Citation2016). The results of Piquereau et al. indicated that mitochondrial autophagy (mitophagy) was activated during sepsis in vivo (Citation2013). Mitophagy is an autophagic response targeting damaged mitochondria specifically, which are isolated via autophagosomes and finally degraded by lysosomes (Turdi et al. Citation2012). Zhong et al. demonstrated the NF-κ b-p62-mitochondrial autophagy pathway, in which damaged mitochondria bind to Parkin-dependent ubiquitination specifically recognized by p62, thus inducing their mitochondrial autophagy clearance (Zhong, Umemura, et al. Citation2016). Studies have also shown that future research on PINK1-Mfn2-Parkin’s mitophagy is instructive (Dorn and Kitsis Citation2015). PARK2/Parkin, an E3 ligase involved in mitophagy regulation in the heart, is important for maintaining the normal mitochondrial function, but the protective role of PARK2/Parkin in mitochondria is only partial (Piquereau et al. Citation2013). Wang’s data suggest that activating FUNDC1-associated mitophagy protects the heart from LPS-induced sepsis by protecting mitochondrial function and structure (Wang et al. Citation2021). Moreover, irisin protected mitochondrial function through FUNDC1-associated mitophagy in LPS-stimulated cardiomyocytes. Zhou et al. also confirmed that FUNDC1-related mitophagy played an important part in cardiac ischemia reperfusion, inflammation-mediated myocardial injury and ischemic preconditioning-afforded cardioprotection (Zhou et al. Citation2018; Wang et al. Citation2021; Zhou et al. Citation2021). There are other related factors (Jiang et al. Citation2021). Shang et al. unearthed that Mst1 deletion had protective influences on mitochondrial homeostasis and cardiomyocyte viability via inducing Parkin-related mitophagy in septic cardiomyopathy (Shang et al. Citation2020). It has been found that increased Mst1 can promote the initiation of mitochondrial apoptosis, and its decrease could activate mitophagy (Shang et al. Citation2020). In addition, mitochondrial respiration and oxidation status and mitochondrial fission are also regulated by Mst1 (Zhou et al. Citation2019). The use of both melatonin and irisin ameliorates LPS-induced cardiac dysfunction via inhibiting the Mst1-JNK pathway, thereby promoting cardiomyocyte survival and mitochondrial homeostasis (Ouyang et al. Citation2020). Although autophagy is essential for the clearance of defective mitochondria, it is also involved in apoptosis and, if not tightly controlled, may lead to cell damage and death, suggesting that a proper understanding of the underlying mechanisms of cardiac autophagy is extremely urgent (Morales et al. Citation2019).
Various studies have shown that the role of autophagy is not limited to clearing the mitochondria of harmful dysfunction within cells but is also involved in regulating inflammatory pathways (Aki et al. Citation2017). Recent research has shown that liraglutide could inhibit NLRP3 inflammasome activation, attenuate mitochondrial dysfunction and ROS generation, and augment mitophagy in hepatocytes, thus suppressing inflammatory injury (Yu et al. Citation2019). Under septic exposure, mitophagy was extensively activated, but impaired mitophagy resulted in overactivation of NLRP3 inflammasomes and an increase in the mortality of septic animals (Kim et al. Citation2016). It was believed it was because autophagy can inhibit inflammatory activation, and autophagosomes could isolate inflammatory body components (Zhong, Sanchez-Lopez, et al. Citation2016). The study found that in sepsis, SESN2-ulk1-mediated mitochondrial selective autophagy is initiated by SESN2-sqstm1, thereby inhibiting NLRP3 inflammasome activation (Kim et al. Citation2016). Therefore, it can be speculated that we can link the NLRP3 inflammasome with the mitophagy for research into the mitochondrial mechanism associated with septic cardiomyopathy. It has already been proved that BMSC (bone marrow stromal cells) could play a beneficial role in inhibiting the activation of macrophage NLRP3 inflammasome by enhancing mitophagy and reducing mitochondrial ROS, thereby combating sepsis (Li et al. Citation2018). Autophagy also plays a role in determining the fate of IL-1β, which is concentrated in autophagosomes (Harris et al. Citation2017). There is also a significant interplay between mitophagy and mitochondrial dynamics. It is concluded that mitochondrial autophagy plays a vital protective role in the synergy with mitochondrial dynamics through the potential interaction between mitochondrial dynamic factors and autophagic receptors (Yao et al. Citation2020). However, we know that mitochondrial fission can increase mitochondrial organelles, which may be a process of mitosis, but it may also be the beginning of apoptosis (Lee et al. Citation2016; Michalska et al. Citation2016; Hasnat et al. Citation2019; Li et al. Citation2020). There are a number of significant interactions between mitophagy and mitochondrial dynamics, which will be discussed in the following paragraphs.
3.2. The NLRP3 inflammasome
The production of mitochondrial ROS can lead to oxidative damage, which in turn leads to further mitochondrial dysfunction as well as the promotion of inflammation (Tsolaki et al. Citation2017). During this process, the NLRP3 inflammasome is activated. Complement activation is a commonly recognized feature of sepsis, particularly involving in the production of C5a and c5b-9 and the binding of c5a-related receptors (Ward and Fattahi Citation2019). There was also evidence that CLP (cecal ligation and puncture) caused mitochondrial dysfunction and spontaneous release of IL-1β and IL-18 by activating NLRP3 inflammasome in the septic heart or in cardiomyocytes incubated in vitro with c55 (Kalbitz et al. Citation2015). Fattahi et al. found that histones cause loss of homeostasis in the redox system, and that Ca2+ system dysfunction may be a target for regulation to reduce septic cardiac dysfunction. It is also said that extracellular histones present in sepsis plasma require C5a receptors, polymorphonuclear leukocytes (PMNs), and NLRP3 inflammasomes (Kalbitz et al. Citation2015; Fattahi et al. Citation2018). It can be speculated that the mitochondrial mechanism and NLRP3 inflammasome associated in septic cardiomyopathy could be linked together. Research shows that, in addition to the classic NF-κB pathway, the presence of the NLRP3/mitochondrial innate immune pathway can also explain the lack of effective treatment strategies for sepsis (Volt et al. Citation2016). Mitochondria and NLRP3 machinery seem to be closely intertwined at multiple levels: for example, ROS production or mtDNA from mitochondrial dysfunction acts upstream of NLRP3 activation; in addition, mitochondria can be used as a platform for inflammasome assembly. Mitochondrial events also contribute downstream to NLRP3 activation, suggesting that NLRP3 inflammasomes can sense mitochondrial dysfunction, which may explain the close relationship between mitochondrial damage and inflammatory disease (Zhou et al. Citation2011; Yu and Lee Citation2016). So, it can be speculated that treatment could be done by targeting NLRP3 inflammasomes. Here are some examples of instantiations. The study of Volt et al. showed that acute treatment of melatonin in aging mice could reduce the over-reaction of the NF-κB/NLRP3 response in the septic heart (Volt et al. Citation2016). Research has also suggested that SO2 can protect the heart from sepsis-induced cardiac dysfunction, which might be associated with the inhibition of inflammation via TLR4/NLRP3 signaling pathway (Yang et al. Citation2018).
3.3. Uncoupling protein 2 (UCP2) in mitochondria
Uncoupling means that the conservation of energy between substrate oxidation and ADP phosphorylation is broken, which would be fatal, but fine-tuning it may help prevent the release of mitochondrial ROS (Bouillaud et al. Citation2016). The coupled or uncoupled state of mitochondria is related to mitochondrial inner membrane permeability and proton transport. UCP2, a family of mitochondrial anionic proteins, is present in the inner mitochondrial membrane (Sreedhar and Zhao Citation2017). There is strong evidence that UCP1 homologs, UCP2 and UCP3, expressed in the heart protect against mitochondrial oxidative damage by reducing ROS production, and it has been suggested that UCP2 and UCP3 have similar functions (Cadenas Citation2018).
UCP2 is the most-studied UCP currently. The data indicates that mitochondrial morphology and function of cardiomyocytes are disrupted under sepsis conditions, and the use of siRNA to silence UCP2 exacerbates this process (Zheng et al. Citation2015). UCP2 is an important mediator of Ca2+ transport, and disruption of Ca2+ delivery due to sustained ER stress or inhibition of UCP2 expression can cause mitochondrial dysfunction (Dromparis et al. Citation2013). There are other findings concerning UCP2 in septic cardiomyopathy. Chen et al. demonstrated that UCP2 overexpression reversed sepsis-induced myocardial damage by modulating mitoflash frequency (Chen et al. Citation2018). Furthermore, earlier study demonstrated that a small heterodimer partner inhibited systemic inflammatory response through mitochondrial UCP2 during endotoxin-induced septic shock (Yang et al. Citation2013). There are also experimental reports that UCP2 prevents renal tubular epithelial cells from apoptosis induced by LPS by reducing the production of excessive ROS (Zhong, He, et al. Citation2019). Moreover, it is said that UCP2 protects against LPS-induced oxidative stress and apoptosis in cardiomyocytes, in which the protein levels of phosphorylated p38 may be related (Huang et al. Citation2019). In addition, a series of studies by Mao et al. showed that UCP2 plays a cardioprotective role in mitochondrial dysfunction during sepsis by regulating the balance of autophagy and apoptosis via the AMPK pathway (Mao et al. Citation2021). Eyenga et al. found that up-regulation of UCP2 mRNA expression led to decreased mitochondrial ROS production at 24 h after CLP onset, and ROS overproduction was accompanied by decreased UCP2 mRNA expression at 36 h (Eyenga et al. Citation2018). Furthermore, correlation analysis showed that UCP2 protein and mRNA levels were negatively correlated with myocardial energy levels during endotoxic shock (Wang et al. Citation2015). Therefore, further studies are needed, which could pave the way for innovative treatment strategy of sepsis with heart failure (Wang et al. Citation2015). Thus, it comes to controversial opinions about UCP2 in septic cardiomyopathy. The results showed that UCP2 regulated and eventually activated NLRP3 inflammasome by inducing the lipid synthesis pathway in macrophages (Moon et al. Citation2015). However, the results can confirm that UCP2 is a potential therapeutic target and specific marker for inflammatory diseases such as sepsis (Moon et al. Citation2015; Jiang et al. Citation2017). Moon et al. demonstrated that autophagy inhibitor 3-MA eliminated the cardioprotective effect produced by UCP2 knockdown, and other results showed that UCP2 drove septic cardiac dysfunction (Tang et al. Citation2019). Roshon et al. claimed that they did not find any evidence of UCP2 protein in septic myocardium. These data contradict the hypothesis that UCP2 contributes to the reduced mechanical efficiency of septic hearts (Roshon et al. Citation2003). We also learned that functional polymorphisms in the promoter region of the UCP2 gene are associated with hyperglycemia and insulin resistance in patients with severe sepsis (Pyle et al. Citation2009). In the ventromedial nucleus of the hypothalamus (VMH), glucose induces mitochondrial fission and dynamin-related protein 1 (Drp1) activation, which is regulated by UCP2 (Toda et al. Citation2016). This raises the possibility of UCP2 being involved in mitochondrial fission in the septic heart. The relationship between UCP2 and septic cardiomyopathy requires further exploration.
3.4. Dynamin-related protein 1 (Drp1) in mitochondrial fission
Mitochondrial dynamics are the first reaction to mitochondrial damage (Sciarretta et al. Citation2018). Studies have shown that mitochondrial dynamics are important for regulating energy demand and mitochondrial homeostasis coordination, which is a biodynamic adaptation mechanism for the physiological reconstruction of sepsis cardiology (Vasquez-Trincado et al. Citation2016). The most studied parts of cardiac mitochondrial dynamics are mitochondrial fission and fusion, biogenesis, and mitochondrial clearance (Morales et al. Citation2019). Fission and fusion can facilitate the exchange of metabolites and substrates between mitochondria, which is an important part of maintaining homeostasis (Youle and Van Der Bliek Citation2012; Friedman and Nunnari Citation2014). Mitochondrial fission characterized by the division of mitochondria into short round organelles, then followed the removal of the damaged part of the mitochondria through mitophagy, thus preserving the healthy part of the mitochondria and maintaining its function (Saito and Sadoshima Citation2015). Mitochondrial fusion involves multiple steps to bind the membranes of adjacent mitochondria for fusion, while fission splits mitochondria into two sub-mitochondria.
Mitochondrial fusion depends on dynein-related GTPases, mitofusin1 (Mfn1) and mitofusin2 (Mfn2) on the outer mitochondrial membrane (OMM) and Opa1 on the inner membrane (Hoppins and Nunnari Citation2009). In ischemic disorders, OPA1 inactivation can lead to obstruction of fusion, fragmentation of mitochondria, and eventually increased apoptosis and organ damage (Xiao et al. Citation2014). Mitochondrial fragmentation may increase Bax (apoptosis-related molecules) ‘attack’ (Parikh et al. Citation2015). Future studies are needed to determine whether this mechanism is applicable to sepsis. Drp1 mediates mitochondrial fission. DRP1 has two phosphorylation sites, Ser616 and Ser637. Phosphorylation of Ser616 promotes oligomerization of Drp1 around OMM, which is a prerequisite for the formation of a potential mitochondrial fission ring (Wang and Zhou Citation2020). Phosphorylation of Ser637 has the opposite effect (Sharp et al. Citation2014). The findings of Shen’s study suggested that the TNF-α caused phosphorylation of Drp1 S616 during inflammatory myocardial injury and mitochondrial fragmentation (Shen et al. Citation2018). Studies have shown that the mitochondrial fission inhibitor Mdivi-1 can reduce cardiac and vascular smooth muscle inflammation and apoptosis induced by sepsis by inhibiting mitochondrial fission and improving mitochondrial function (Zhu et al. Citation2021). Elimination of Drp1 in adult mouse cardiomyocytes not only interferes with mitochondrial fission, but also significantly up-regulates Parkin, causing mitophagy depletion, leading to fatal cardiomyopathy (Song et al. Citation2015). This implies that Drp1 is indispensable in the cardiomyocytes. However, it has been shown that inhibition of the Drp1/Fis1 interaction could attenuate pathological fission and subsequently mitochondrial dysfunction, thereby improving cardiac function and increasing sepsis survival (Haileselassie et al. Citation2019). Shang et al. defined the Mst1/Drp1/mitochondrial fission/F-actin axis as a new signaling pathway that mediates LPS-related septic cardiomyopathy by inducing mitochondrial stress and cardiomyocyte death (Shang et al. Citation2019). This gives us an indication that we should find out exactly how much mitochondrial fission is appropriate. We should also work out how to treat this, and melatonin might be a good idea. It is suggested that melatonin-mediated inhibition of receptor-interacting protein kinase (Ripk3) improves mitochondrial biogenesis, sustains mitochondrial dynamics, reduces ER stress, normalizes Ca2+ circulation, and activates protection of cardiomyocyte signal pathways (including AKT, ERK and AMPK) (Zhong, Tan, et al. Citation2019). The results of Tan et al. indicated that LPS-mediated Drp1-related mitochondrial fission activated by the JNK-LATS2 pathway was involved in myocardial suppression caused by sepsis, and irisin may be an effective therapy in the future (Tan et al. Citation2019). In addition to Drp1, mitochondrial fission is regulated by JNK pathway. Activation of the latter suppresses the mitochondrial fission factor (Mff)-required mitochondrial fission and Bnip3-related mitophagy (Jin et al. Citation2018). The Mff-dependent mitochondrial fission contributes to the mPTP openings in a mechanism involving induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 dissociation (Zhou et al. Citation2017).
3.5. Peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) in mitochondrial biogenesis
Mitochondrial biogenesis refers to the proliferation of mitochondria mass and the systematic and individual synthesis of mitochondria DNA during the life cycle of a cell. It is essential for renewing damaged mitochondria and increasing energy production (Youle and Van Der Bliek Citation2012). During inflammation, persistent oxidative stress can initiate retrograde signaling, activating mitochondrial biogenesis (Piantadosi and Suliman Citation2012). However, pathological acceleration of mitochondrial biogenesis may indicate inadequate mitochondrial function (Russell et al. Citation2004). There is still untapped therapeutic potential for drugs that activate mitochondrial biogenesis and related metabolic pathways in addressing dangerous immune responses to severe infections (Fedson Citation2009).
It is believed that PGC-1α, nuclear respiratory factor-1 (NRF-1), and mitochondrial transcription factor-A (TFAM) play key regulatory roles in mitochondrial biogenesis (Scarpulla Citation2008; Carre et al. Citation2010; Gureev et al. Citation2019). NRF-1 can be activated by LPS and promote mitochondrial biogenesis through TLR4 mechanism to participate in the host antibacterial defenses (Suliman et al. Citation2010). TFAM was also essential to increase mitochondrial gene transcription when the mtDNA was damaged in sepsis.
PGC-1α and related transcription coactivators are major transcriptional regulators of mitochondrial biogenesis in sepsis (Arany et al. Citation2005). PGC-1α can be activated in response to growth signals or lack of energy (Handschin and Spiegelman Citation2006). Targets of PGC-1α include energy metabolism enzymes and factors are necessary for mtDNA replication and transcription (Scarpulla Citation2011). The regulatory role of PGC-1α in pathogenesis has been demonstrated in different animal models (Arany et al. Citation2005; Kulikova et al. Citation2018). Nevertheless, increasing PGC-1α levels as a therapeutic strategy has often encountered conflicting results (Lehman et al. Citation2000; Russell et al. Citation2004; Finck and Kelly Citation2007). In a recent study, Zhu et al. used a strategy of fine-tuning the expression of PGC-1α, which turned out to be essential for cardiac homeostasis (Zhu et al. Citation2019). Their experimental results suggest the importance of carefully evaluating PGC-1α enhancement strategies and facilitating the clinical translation of new cardiovascular therapies (Zhu et al. Citation2019). However, biogenesis does not seem to necessarily equate to the restoration of mitochondrial function. Vanasco’s results showed that myocardial mitochondrial biogenesis during endotoxemia was not accompanied by mitochondrial function recovery (Vanasco et al. Citation2014). Moreover, mitochondrial oxygen consumption and respiratory control rate (RCR), as well as mitochondrial complex I activity, did not return to normal values when mitochondrial biomarkers were significantly increased after LPS treatment (Vanasco et al. Citation2014). Suliman et al. have shown that the level of PGC-1α increased 6 h after LPS treatment, and the expression levels of complexes I and IV decreased 24 h after LPS treatment (50–30%, respectively) (Suliman et al. Citation2004). Therefore, further study of biogenesis is needed to better understand its effects on mitochondrial and organ function.
Yuan et al. found that autophagy was activated prior to mitochondrial biogenesis (Yuan et al. Citation2009). The role of autophagy is still controversial. Thus, the final state of the septic heart depends on complex relationships between different processes, which mainly include mitochondrial biogenesis, autophagy, and cell death (Alvarez et al. Citation2016). These processes are still worth exploring in the future. Zhang et al. concluded that mechanistic target of rapamycin (mTOR) pathway, the local translation of mitochondrial proteins on the outer membrane, mitochondrial DNA insufficient-La-related protein (MDI–Larp) complex and some miRNAs (like miR-494, miR-338, miR-210) could regulate mitochondrial biogenesis (Zhang and Xu Citation2016). Ryter discussed that heme oxygenase-1 (HO-1) might be localized to mitochondria in response to stress (Lancel et al. Citation2009), while carbon monoxide (CO) might relieve mitochondrial dysfunction and regulate mitophagy and mitochondrial biogenesis in sepsis and/or septic cardiomyopathy (Lancel et al. Citation2009; Wang et al. Citation2014; Ryter Citation2019).
3.6. The Ca2 + and MAMs involved in mitochondria
Mitochondria are the site of intracellular Ca2+ accumulation and the main site of intercellular Ca2+ balance and regulation (Zhang et al. Citation2018). Mitochondrial Ca2+ overload is an important mechanism responsible for mitochondrial damage in sepsis. Mitochondrial Ca2+ overload can impede ATP synthesis, which then triggers the irreversible prolonged opening of mPTP. However, excessive opening of mPTP can lead to the release of cytochrome c, following with caspase activation, and eventually lead to cell death and cardiomyocyte contractile dysfunction (Yarana et al. Citation2012; Kwong and Molkentin Citation2015).
The instantaneous opening of Ca2+-dependent mPTP may provide a fast Ca2 + release channel for mitochondria, preventing Ca2+ overload (Bernardi and Von Stockum Citation2012). Elrod et al. confirmed that CypD and mPTP are important regulators of mitochondrial Ca2+ exchange in cardiomyocytes and are essential for maintaining metabolic adaptation (Elrod et al. Citation2010). There is no doubt that maintaining mitochondrial Ca2+ homeostasis by mPTP is essential for the septic heart. However, its connection to other functions of mitochondria is still worth investigating. It is suggested that Ca2+/calmodulin-dependent protein kinase (CaMK) linked mitochondrial stress with the PINK1/Parkin and DJ-1 pathway of mitophagy in sepsis (Zhang, Yuan, et al. Citation2017). Moreover, mitochondrial Ca2+ overload can overproduce ROS, which in turn promotes Ca2+ overload (Morales et al. Citation2019). Therefore, reducing ROS is also important. Joseph et al. revealed that inhibition of NADPH oxidase 2 (NOX2) prevented sepsis-induced cardiomyopathy by improving Ca2+ handling and mitochondrial function (Joseph et al. Citation2017).
The results show that infectious cardiomyopathy can activate ER stress, and melatonin reduces ER stress through Ripk3, thereby protecting Ca2+ homeostasis (Zhong, Tan, et al. Citation2019). The ER can form stable contact sites with many other cytoplasmic organelles, including the mitochondria, for functional collaboration (Rowland and Voeltz Citation2012). A section of the ER communicating with mitochondria is referred to as the mitochondria-associated membranes (MAMs) (Rowland and Voeltz Citation2012). MAMs consist of a protein tether that physically connects ER to mitochondria (Van Vliet and Agostinis Citation2018). MAMs are critical for the formation of intracellular Ca2+ signaling and the regulation of proper mitochondrial bioenergetics and mitochondrial dynamics (Szabadkai et al. Citation2006; Wu et al. Citation2017; Van Vliet and Agostinis Citation2018). Wu et al. concluded that the binding of FUN14 domain containing 1 (FUNDC1) with inositol 1,4,5-trisphosphate receptor (INSP3R) in mitochondria-associated ER membranes maintained mitochondrial dynamics in the heart (Wu et al. Citation2017). Furthermore, the FUNDC1 binds to inositol 1,4,5-trisphosphate type 2 receptor (IP3R2) to modulate ER Ca2+ release into mitochondria and cytosol, the destruction of which leads to impaired mitochondrial dynamics of cardiomyocytes (Wu et al. Citation2017). At the molecular level, the presence of inositol requiring-enzyme 1 alpha (IRE1α) at MAMs determines the availability of INSP3R and therefore facilitates Ca2+ transfer to promote mitochondrial respiration and ATP production (Carreras-Sureda et al. Citation2019). In summary, results indicate that IRE1α expressed at MAMs regulated cellular bioenergy by fine-tuning the communication between ER and mitochondria (Carreras-Sureda et al. Citation2019). It is also suggested that ER MAMs surround mitochondria and bind to the Drp1 protein on OMM to trigger mitochondrial fission in some way (Friedman et al. Citation2011). Wu et al. defined FUNDC1 as the molecular hub where MAMs integrated mitochondrial fission and mitophagy in response to hypoxia (Wu et al. Citation2016). In addition, the production of cardiac ATP mainly depends on oxidative phosphorylation in mitochondria and is dynamically regulated by Ca2+ levels in the mitochondrial matrix and cytosolic ADP (Boyman et al. Citation2019). Boyman et al. propose that the physiological role of MAMs as well as mitochondrial quality control and ATP production needs to be quantitatively defined (Citation2019). MAMs interface can also function as a molecular platform for inflammasome formation and form a key site for autophagosome assembly (Dorn and Kitsis Citation2015). For example, it has been reported that NLRP3 could directly bind to the MAMs and be activated by MAMs-derived effectors (Zhang, Meszaros, et al. Citation2017). It is suggested that, after inhibition of ER-mitochondrial Ca2+ transfer, AMPK presented at MAMs activated local autophagy through beclin 1 (BECN1) (Ahumada-Castro et al. Citation2019). This local reaction may prevent autophagy-derived metabolites from reaching anabolic pathways, which can cause a homeostatic imbalance (Ahumada-Castro et al. Citation2019).
Taken together, MAMs can form important intracellular signaling platforms that allow for the exchange of information between ER and mitochondria (Kerkhofs et al. Citation2018). The crosstalk between mitochondria and ER is a hot topic in biological research, and their molecular communication and their multitudinous roles in multiple signaling pathways are constantly evolving (Rieusset Citation2018). For this reason, it seems crucial to clarify the precise knowledge of MAMs molecules and their molecular mechanisms controlling interorganelle communication (Bernardi and Von Stockum Citation2012; Veeresh et al. Citation2019). The research of MAMs in cardiology will also be a hot spot from now to the future, and it may suggest novel targets in the setting of septic cardiomyopathy (Naon and Scorrano Citation2014; Van Vliet et al. Citation2014; Wu et al. Citation2019).
4. Conclusion
Cardiac insufficiency due to sepsis is significantly associated with sepsis mortality. However, the exact mechanism of septic cardiomyopathy remains unclear. Studies have reported reduced cardiac mitochondrial content and mitophagy in animal models of sepsis. Clinical investigations have also shown that the severity of mitochondrial dysfunction is closely related to the prognosis of sepsis and cardiac dysfunction. It is reasonable to believe that mitochondrial dysfunction plays an essential part in septic cardiomyopathy.
This review summarizes the potential mechanisms of mitochondrial dysfunction in septic cardiomyopathy. These mechanisms include mitophagy and sepsis-induced inflammation pathway, mitochondrial uncoupling, mitochondrial fission and fusion, mitochondrial biogenesis and mitochondrial processing of Ca2+. We have observed that mitophagy is used to clear damaged mitochondria, but it is well known that there remains some controversy about the role autophagy is playing in the body. Therefore, must mitochondrial autophagy be protective against cardiomyopathy during sepsis? This is waiting for the discovery of more molecular mechanisms for further investigation and exploration.
This review also focuses on the possible interaction between mitochondrial components and NLRP3 inflammasome in myocardial cells. Stimulating factors such as LPS and ATP can promote mitochondrial dysfunction, generation of ROS and damaged mtDNA, which are activators of NLRP3 inflammasome in the cytoplasm. Therefore, NLRP3 inflammasome is finally activated and exerts its effects. During this process, mitochondria and its associated membranes can provide a platform for inflammasome assembly. However, the effect of autophagy is to isolate the components of the inflammasome and prevent its activation.
A better understanding of the interaction between the inflammasome and the autophagosome could lead to new therapeutic targets. There are some molecules, such as the UCP2 in mitochondrial uncoupling, the PGC-1α in biogenesis, the FUNDC1 involved in MAMs and Ca2+ processing, and the Drp1 in mitochondrial dynamics, all of which are hot spots to be thoroughly studied in the future. Further studies of the activity and regulation of MAMs could improve our understanding of myocardial depression. More research is needed to develop new drugs or promising therapies that may be used to reverse mitochondrial damage associated with multiple mechanisms.
In the meantime, there appears to be the emergence of a number of therapeutic targets for mitochondrial dysfunction associated with septic cardiomyopathy. Among these, carbon monoxide therapy and treatment with melatonin are the most compelling. Certainly, further studies are needed to explore their specific mechanisms before these treatments can be promoted. It is believed that more clear mechanisms will be displayed to help people better understand and treat septic cardiomyopathy with the rapid development of novel scientific research technology and the unremitting efforts of scientific researchers.
Acknowledgements
Dr Yuting Tang, and Dr Xiaofang Lin performed literature searches. Prof. Bimei Jiang revised the review. The first draft of the manuscript and the graphical abstract were written or drawn by Dr Leijing Yin. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Ahumada-Castro U, Silva-Pavez E, Lovy A, Pardo E, Molgomicron J, Cardenas C. 2019. MTOR-independent autophagy induced by interrupted endoplasmic reticulum-mitochondrial Ca(2+) communication: a dead end in cancer cells. Autophagy. 15(2):358–361.
- Aki T, Unuma K, Uemura K. 2017. Emerging roles of mitochondria and autophagy in liver injury during sepsis. Cell Stress. 1(2):79–89.
- Alvarez S, Vico T, Vanasco V. 2016. Cardiac dysfunction, mitochondrial architecture, energy production, and inflammatory pathways: interrelated aspects in endotoxemia and sepsis. Int J Biochem Cell Biol. 81(Pt B):307–314.
- Anderson DP, Allen WJ, Barcroft H, Edholm OG, Manning GW. 1946. Circulatory changes during fainting and coma caused by oxygen lack. J Physiol. 104(4):426–434.
- Antonucci E, Fiaccadori E, Donadello K, Taccone FS, Franchi F, Scolletta S. 2014. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J Crit Care. 29(4):500–511.
- Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH , et al. 2005. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 1(4):259–271.
- Beesley SJ, Weber G, Sarge T, et al. 2018. Septic cardiomyopathy. Crit Care Med. 46(4):625–634.
- Belcher E, Mitchell J, Evans T. 2002. Myocardial dysfunction in sepsis: no role for NO? Heart. 87(6):507–509.
- Bernardi P, Von Stockum S. 2012. The permeability transition pore as a Ca(2+) release channel: new answers to an old question. Cell Calcium. 52(1):22–27.
- Bouillaud F, Alves-Guerra M C, Ricquier D. 2016. UCPs, at the interface between bioenergetics and metabolism. Biochim Biophys Acta. 1863(10):2443–2456.
- Boyman L, Karbowski M, Lederer WJ. 2019. Regulation of mitochondrial ATP production: Ca(2+) signaling and quality control. Trends Mol Med. 26(1):21–39.
- Bravo-San Pedro JM, Kroemer G, Galluzzi L. 2017. Autophagy and mitophagy in cardiovascular disease. Circ Res. 120(11):1812–1824.
- Brealey D, Brand M, Hargreaves I, et al. 2002. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 360(9328):219–223.
- Bruni FD, Komwatana P, Soulsby ME, et al. 1978. Endotoxin and myocardial failure: role of the myofibril and venous return. Am J Physiol. 235(2):H150–H156.
- Cadenas S. 2018. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg. 1859(9):940–950.
- Cao F, Maguire ML, Mcandrew DJ, et al. 2020. Overexpression of mitochondrial creatine kinase preserves cardiac energetics without ameliorating murine chronic heart failure. Basic Res Cardiol. 115(2):12.
- Carre JE, Orban JC, Re L, et al. 2010. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 182(6):745–751.
- Carreras-Sureda A, Jana F, Urra H, et al. 2019. Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat Cell Biol. 21(6):755–767.
- Chen W, Luo S, Xie P, et al. 2018. Overexpressed UCP2 regulates mitochondrial flashes and reverses lipopolysaccharide-induced cardiomyocytes injury. Am J Transl Res. 10(5):1347–1356.
- Cimolai MC, Alvarez S, Bode C, et al. 2015. Mitochondrial mechanisms in septic cardiomyopathy. Int J Mol Sci. 16(8):17763–17778.
- Coopersmith CM, DE BACKER D, Deutschman CS, et al. 2018. Surviving sepsis campaign: research priorities for sepsis and septic shock. Crit Care Med. 46(8):1334–1356.
- Dorn GW 2nd, Kitsis RN. 2015. The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res. 116(1):167–182.
- Dromparis P, Paulin R, Sutendra G, et al. 2013. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res. 113(2):126–136.
- Durand A, Duburcq T, Dekeyser T, et al. 2017. Involvement of mitochondrial disorders in septic cardiomyopathy. Oxid Med Cell Longev. 2017:4076348.
- Ehrman RR, Sullivan AN, Favot MJ, et al. 2018. Pathophysiology, echocardiographic evaluation, biomarker findings, and prognostic implications of septic cardiomyopathy: a review of the literature. Crit Care. 22(1):112.
- Elrod JW, Wong R, Mishra S, et al. 2010. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 120(10):3680–3687.
- Escames G, Lopez LC, Tapias V, et al. 2006. Melatonin counteracts inducible mitochondrial nitric oxide synthase-dependent mitochondrial dysfunction in skeletal muscle of septic mice. J Pineal Res. 40(1):71–78.
- Eyenga P, Roussel D, Morel J, et al. 2018. Time course of liver mitochondrial function and intrinsic changes in oxidative phosphorylation in a rat model of sepsis. Intensive Care Med Exp. 6(1):31.
- Fang X, Wang J. 2018. [Role of mitochondrial dysfunction in the pathogenesis of septic cardiomyopathy]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 30(2):189–192.
- Fattahi F, Frydrych LM, Bian G, et al. 2018. Role of complement C5a and histones in septic cardiomyopathy. Mol Immunol. 102:32–41.
- Fedson DS. 2009. Meeting the challenge of influenza pandemic preparedness in developing countries. Emerg Infect Dis. 15(3):365–371.
- Fender AC, Kleeschulte S, Stolte S, et al. 2020. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res Cardiol. 115(2):10.
- Finck BN, Kelly DP. 2007. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 115(19):2540–2548.
- Fink MP. 2001. Cytopathic hypoxia. mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 17(1):219–237.
- Fink MP, Warren HS. 2014. Strategies to improve drug development for sepsis. Nat Rev Drug Discov. 13(10):741–758.
- Friedman JR, Lackner LL, West M, et al. 2011. ER tubules mark sites of mitochondrial division. Science. 334(6054):358–362.
- Friedman JR, Nunnari J. 2014. Mitochondrial form and function. Nature. 505(7483):335–343.
- Gureev AP, Shaforostova EA, Popov VN. 2019. Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1alpha signaling pathways. Front Genet. 10:435.
- Haileselassie B, Mukherjee R, Joshi AU, et al. 2019. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol. 130:160–169.
- Handschin C, Spiegelman BM. 2006. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 27(7):728–735.
- Hardeland R. 2018. Melatonin and inflammation-story of a double-edged blade. J Pineal Res. 65(4):e12525.
- Harris J, Lang T, Thomas JPW, et al. 2017. Autophagy and inflammasomes. Mol Immunol. 86:10–15.
- Hasnat M, Yuan Z, Naveed M, et al. 2019. Drp1-associated mitochondrial dysfunction and mitochondrial autophagy: a novel mechanism in triptolide-induced hepatotoxicity. Cell Biol Toxicol. 35(3):267–280.
- Hoppins S, Nunnari J. 2009. The molecular mechanism of mitochondrial fusion. Biochim Biophys Acta. 1793(1):20–26.
- Horak J, Martinkova V, Radej J, et al. 2019. Back to basics: recognition of sepsis with new definition. J Clin Med. 8(11):1838.
- Hotchkiss RS, Karl IE. 1992. Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis. Jama. 267(11):1503–1510.
- Huang J, Peng W, Zheng Y, et al. 2019. Upregulation of UCP2 expression protects against LPS-induced oxidative stress and apoptosis in cardiomyocytes. Oxid Med Cell Longev. 2019:2758262.
- Jiang BM, Liang PF, Tang YT, et al. 2019. [Study on the expression and roles of nucleolin in cardiac injury in septic mice]. Zhonghua yi xue za zhi. 99(1):57–61.
- Jiang X, Cai S, Jin Y, et al. 2021. Irisin attenuates oxidative stress, mitochondrial dysfunction, and apoptosis in the H9C2 cellular model of septic cardiomyopathy through augmenting Fundc1-dependent mitophagy. Oxid Med Cell Longev. 2021:2989974.
- Jiang ZM, Yang QH, Zhu CQ. 2017. UCP2 in early diagnosis and prognosis of sepsis. Eur Rev Med Pharmacol Sci. 21(3):549–553.
- Jin Q, Li R, Hu N, et al. 2018. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 14:76–87.
- Joseph LC, Kokkinaki D, Valenti MC, et al. 2017. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2(17):e94248.
- Kalbitz M, Grailer JJ, Fattahi F, et al. 2015. Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J. 29(5):2185–2193.
- Kerkhofs M, Bittremieux M, Morciano G, et al. 2018. Emerging molecular mechanisms in chemotherapy: Ca(2+) signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 9(3):334.
- Kim MJ, Bae SH, Ryu JC, et al. 2016. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 12(8):1272–1291.
- Kulikova TG, Stepanova OV, Voronova AD, et al. 2018. Pathological remodeling of the myocardium in chronic heart failure: role of PGC-1alpha. Bull Exp Biol Med. 164(6):794–797.
- Kwong JQ, Molkentin JD. 2015. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 21(2):206–214.
- Lancel S, Hassoun S M, Favory R, et al. 2009. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J Pharmacol Exp Ther. 329(2):641–648.
- Lee JE, Westrate LM, Wu H, et al. 2016. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 540(7631):139–143.
- Lehman JJ, Barger PM, Kovacs A, et al. 2000. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 106(7):847–856.
- Lemasters JJ, Theruvath TP, Zhong Z, et al. 2009. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 1787(11):1395–1401.
- Levy RJ. 2007. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock. 28(1):24–28.
- Li J, Zhang B, Chang X, et al. 2020. Silver nanoparticles modulate mitochondrial dynamics and biogenesis in HepG2 cells. Environ Pollut. 256:113430.
- Li S, Wu H, Han D, et al. 2018. A novel mechanism of mesenchymal stromal cell-mediated protection against sepsis: restricting inflammasome activation in macrophages by increasing mitophagy and decreasing mitochondrial ROS. Oxid Med Cell Longev. 2018:3537609.
- Li Y, Feng YF, Liu XT, et al. 2021. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 38:101771.
- Mao JY, Su LX, Li DK, et al. 2021. The effects of UCP2 on autophagy through the AMPK signaling pathway in septic cardiomyopathy and the underlying mechanism. Ann Transl Med. 9(3):259.
- Michalska B, Duszynski J, Szymanski J. 2016. [Mechanism of mitochondrial fission - structure and function of Drp1 protein]. Postepy Biochem. 62(2):127–137.
- Moncada S, Erusalimsky JD. 2002. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol. 3(3):214–220.
- Moon JS, Lee S, Park MA, et al. 2015. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 125(2):665–680.
- Morales PE, Arias-Duran C, Avalos-Guajardo Y, et al. 2019. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med. 71:100822.
- Naon D, Scorrano L. 2014. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim Biophys Acta. 1843(10):2184–2194.
- Ouyang H, Li Q, Zhong J, et al. 2020. Combination of melatonin and irisin ameliorates lipopolysaccharide-induced cardiac dysfunction through suppressing the Mst1-JNK pathways. J Cell Physiol. 235(10):6647–6659.
- Pan P, Wang X, Liu D. 2018. The potential mechanism of mitochondrial dysfunction in septic cardiomyopathy. J Int Med Res. 46(6):2157–2169.
- Pan P, Zhang H, Su L, et al. 2018. Melatonin balance the autophagy and apoptosis by regulating UCP2 in the LPS-induced cardiomyopathy. Molecules. 23(3):675 .
- Parikh SM, Yang Y, He L, et al. 2015. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol. 35(1):108–119.
- Piantadosi CA, Suliman HB. 2012. Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim Biophys Acta. 1820(4):532–541.
- Piel DA, Deutschman CS, Levy RJ. 2008. Exogenous cytochrome C restores myocardial cytochrome oxidase activity into the late phase of sepsis. Shock. 29(5):612–616.
- Piquereau J, Godin R, Deschenes S, et al. 2013. Protective role of PARK2/parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy. 9(11):1837–1851.
- Poggi C, Dani C. 2018. Sepsis and oxidative stress in the newborn: from pathogenesis to novel therapeutic targets. Oxid Med Cell Longev. 2018:9390140.
- Pyle A, Ibbett I M, Gordon C, et al. 2009. A common UCP2 polymorphism predisposes to stress hyperglycaemia in severe sepsis. J Med Genet. 46(11):773–775.
- Qiao Y, Wang L, Hu T, et al. 2021. Capsaicin protects cardiomyocytes against lipopolysaccharide-induced damage via 14-3-3γ-mediated autophagy augmentation. Front Pharmacol. 12:659015.
- Rieusset J. 2018. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis. 9(3):388.
- Roshon MJ, Kline JA, Thornton LR, et al. 2003. Cardiac UCP2 expression and myocardial oxidative metabolism during acute septic shock in the rat. Shock. 19(6):570–576.
- Rowland AA, Voeltz GK. 2012. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 13(10):607–625.
- Russell LK, Mansfield CM, Lehman JJ, et al. 2004. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 94(4):525–533.
- Ryter SW. 2019. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch Biochem Biophys. 678:108186.
- Saito T, Sadoshima J. 2015. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 116(8):1477–1490.
- Sato R, Kuriyama A, Takada T, et al. 2016. Prevalence and risk factors of sepsis-induced cardiomyopathy: a retrospective cohort study. Medicine (Baltimore). 95(39):e5031.
- Scarpulla RC. 2008. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci. 1147:321–334.
- Scarpulla RC. 2011. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 1813(7):1269–1278.
- Sciarretta S, Maejima Y, Zablocki D, et al. 2018. The role of autophagy in the heart. Annu Rev Physiol. 80:1–26.
- Shang X, Li J, Yu R, et al. 2019. Sepsis-related myocardial injury is associated with Mst1 upregulation, mitochondrial dysfunction and the Drp1/F-actin signaling pathway. J Mol Histol. 50(2):91–103.
- Shang X, Lin K, Zhang Y, et al. 2020. Mst1 deletion reduces septic cardiomyopathy via activating parkin-related mitophagy. J Cell Physiol. 235(1):317–327.
- Sharp WW, Fang YH, Han M, et al. 2014. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. Faseb J. 28(1):316–326.
- Shen YL, Shi Y Z, Chen GG, et al. 2018. TNF-α induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int J Mol Med. 41(4):2317–2327.
- Singer M. 2014. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 5(1):66–72.
- Singer M, Deutschman CS, Seymour CW, et al. 2016. The third international consensus definitions for sepsis and septic shock (sepsis-3). Jama. 315(8):801–810.
- Song M, Gong G, Burelle Y, et al. 2015. Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res. 117(4):346–351.
- Sreedhar A, Zhao Y. 2017. Uncoupling protein 2 and metabolic diseases. Mitochondrion. 34:135–140.
- Suliman HB, Sweeney TE, Withers CM, et al. 2010. Co-regulation of nuclear respiratory factor-1 by NFkappaB and CREB links LPS-induced inflammation to mitochondrial biogenesis. J Cell Sci. 123(Pt 15):2565–2575.
- Suliman HB, Welty-Wolf KE, Carraway M, et al. 2004. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res. 64(2):279–288.
- Szabadkai G, Bianchi K, Varnai P, et al. 2006. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2 + channels. J Cell Biol. 175(6):901–911.
- Takasu O, Gaut JP, Watanabe E, et al. 2013. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 187(5):509–517.
- Tan Y, Ouyang H, Xiao X, et al. 2019. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones. 24(3):595–608.
- Tang R, Qi PP, Liu YS, et al. 2019. Uncoupling protein 2 drives myocardial dysfunction in murine models of septic shock. Biomed Res Int. 2019:9786101.
- Toda C, Kim JD, Impellizzeri D, et al. 2016. UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell. 164(5):872–883.
- Trumbeckaite S, Opalka JR, Neuhof C, et al. 2001. Different sensitivity of rabbit heart and skeletal muscle to endotoxin-induced impairment of mitochondrial function. Eur J Biochem. 268(5):1422–1429.
- Tsolaki V, Makris D, Mantzarlis K, et al. 2017. Sepsis-induced cardiomyopathy: oxidative implications in the initiation and resolution of the damage. Oxid Med Cell Longev. 2017:7393525.
- Turdi S, Han X, Huff AF, et al. 2012. Cardiac-specific overexpression of catalase attenuates lipopolysaccharide-induced myocardial contractile dysfunction: role of autophagy. Free Radic Biol Med. 53(6):1327–1338.
- Vanasco V, Saez T, Magnani ND, et al. 2014. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radical Biol Med. 77:1–9.
- Van Vliet AR, Agostinis P. 2018. Mitochondria-associated membranes and ER stress. Curr Top Microbiol Immunol. 414:73–102.
- Van Vliet AR, Verfaillie T, Agostinis P. 2014. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 1843(10):2253–2262.
- Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, et al. 2016. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 594(3):509–525.
- Veeresh P, Kaur H, Sarmah D, et al. 2019. Endoplasmic reticulum-mitochondria crosstalk: from junction to function across neurological disorders. Ann N Y Acad Sci. 1457(1):41–60.
- Volt H, García JA, Doerrier C, et al. 2016. Same molecule but different expression: aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J Pineal Res. 60(2):193–205.
- Wang GQ, Tang T, Wang ZS, et al. 2016. Overexpression of hypo-phosphorylated IkappaBbeta at Ser313 protects the heart against sepsis. PLoS One. 11(8):e0160860.
- Wang J, Zhou H. 2020. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharmaceutica Sinica B. 10(10):1866–1879.
- Wang X, Liu D, Chai W, et al. 2015. The role of uncoupling protein 2 during myocardial dysfunction in a canine model of endotoxin shock. Shock. 43(3):292–297.
- Wang X, Qin W, Qiu X, et al. 2014. A novel role of exogenous carbon monoxide on protecting cardiac function and improving survival against sepsis via mitochondrial energetic metabolism pathway. Int J Biol Sci. 10(7):777–788.
- Wang Y, Jasper H, Toan S, et al. 2021. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 45:102049.
- Ward PA, Fattahi F. 2019. New strategies for treatment of infectious sepsis. J Leukocyte Biol. 106(1):187–192.
- Wu S, Lu Q, Ding Y, et al. 2019. Hyperglycemia-driven inhibition of AMP-activated protein kinase alpha2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation. 139(16):1913–1936.
- Wu S, Lu Q, Wang Q, et al. 2017. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 136(23):2248–2266.
- Wu W, Li W, Chen H, et al. 2016. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. 12(9):1675–1676.
- Xiao X, Hu Y, Quiros PM, et al. 2014. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am J Physiol Renal Physiol. 306(11):F1318–F1326.
- Yang CS, Yuk JM, Kim JJ, et al. 2013. Small heterodimer partner-targeting therapy inhibits systemic inflammatory responses through mitochondrial uncoupling protein 2. PLoS One. 8(5):e63435.
- Yang L, Zhang H, Chen P. 2018. Sulfur dioxide attenuates sepsis-induced cardiac dysfunction via inhibition of NLRP3 inflammasome activation in rats. Nitric Oxide. 81:11–20.
- Yao RQ, Ren C, Xia ZF, et al. 2020. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 17(2):385–401.
- Yarana C, Sripetchwandee J, Sanit J, et al. 2012. Calcium-induced cardiac mitochondrial dysfunction is predominantly mediated by cyclosporine A-dependent mitochondrial permeability transition pore. Arch Med Res. 43(5):333–338.
- Youle RJ, Van Der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science. 337(6098):1062–1065.
- Yu JW, Lee MS. 2016. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res. 39(11):1503–1518.
- Yu X, Hao M, Liu Y, et al. 2019. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur J Pharmacol. 864:172715.
- Yuan H, Perry CN, Huang C, et al. 2009. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 296(2):H470–H479.
- Zhang H, Feng YW, Yao YM. 2018. Potential therapy strategy: targeting mitochondrial dysfunction in sepsis. Mil Med Res. 5(1):41.
- Zhang J, Wang L, Xie W, et al. 2020. Melatonin attenuates ER stress and mitochondrial damage in septic cardiomyopathy: A new mechanism involving BAP31 upregulation and MAPK-ERK pathway. J Cell Physiol. 235(3):2847–2856.
- Zhang X, Yuan D, Sun Q, et al. 2017. Calcium/calmodulin-dependent protein kinase regulates the PINK1/Parkin and DJ-1 pathways of mitophagy during sepsis. Faseb J. 31(10):4382–4395.
- Zhang Y, Xu H. 2016. Translational regulation of mitochondrial biogenesis. Biochem Soc Trans. 44(6):1717–1724.
- Zhang Z, Meszaros G, He WT, et al. 2017. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med. 214(9):2671–2693.
- Zheng G, Lyu J, Liu S, et al. 2015. Silencing of uncoupling protein 2 by small interfering RNA aggravates mitochondrial dysfunction in cardiomyocytes under septic conditions. Int J Mol Med. 35(6):1525–1536.
- Zhong J, Tan Y, Lu J, et al. 2019. Therapeutic contribution of melatonin to the treatment of septic cardiomyopathy: A novel mechanism linking Ripk3-modified mitochondrial performance and endoplasmic reticulum function. Redox Biol. 26:101287.
- Zhong X, He J, Zhang X, et al. 2019. UCP2 alleviates tubular epithelial cell apoptosis in lipopolysaccharide-induced acute kidney injury by decreasing ROS production. Biomed Pharmacother. 115:108914.
- Zhong Z, Sanchez-Lopez E, Karin M. 2016. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol. 34(4 Suppl 98):12–16.
- Zhong Z, Umemura A, Sanchez-Lopez E, et al. 2016. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 164(5):896–910.
- Zhou H, Hu S, Jin Q, et al. 2017. Mff-Dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc. 6(3):e005328.
- Zhou H, Ren J, Toan S, et al. 2021. Role of mitochondrial quality surveillance in myocardial infarction: from bench to bedside. Ageing Res Rev. 66:101250.
- Zhou H, Zhu P, Wang J, et al. 2018. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 25(6):1080–1093.
- Zhou R, Yazdi AS, Menu P, et al. 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature. 469(7329):221–225.
- Zhou T, Chang L, Luo Y, et al. 2019. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 21:101120.
- Zhu X, Shen W, Yao K, et al. 2019. Fine-tuning of PGC1alpha expression regulates cardiac function and longevity. Circ Res. 125(7):707–719.
- Zhu Y, Kuang L, Wu Y, et al. 2021. Protective effects of inhibition of mitochondrial fission on organ function after sepsis. Front Pharmacol. 12:712489.
- Zorov DB, Juhaszova M, Sollott SJ. 2014. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 94(3):909–950.