
Abstract
As a regulatory cell death caused by iron-dependent lipid peroxidation, ferroptosis is very important in the healthy development or disease progression of various organisms. Dexamethasone is a glucocorticoid with vigorous immunosuppressive activity and anti-inflammatory effects. On the other hand, it can cause some side effects under long-term or high-dose administration, including secondary osteoporosis and osteonecrosis, which are associated with the elevated autophagy and involvement of ferroptosis. Autophagy, an intracellular degradation system, plays a crucial role in ferroptosis. It has been increasingly proved that ferroptosis is potentially related to the dexamethasone-induced side effects, in which autophagy may be involved. However, the regulatory mechanism of ferroptosis in the dexamethasone-induced side effects is unclear. This review summarizes the molecular mechanisms of ferroptosis, the dexamethasone-induced side effects, and the potential relationships between ferroptosis and dexamethasone-induced side effects, aiming to provide novel insight into the interplay between them.
Introduction
Ferroptosis refers to a regulatory cell death caused by impairment of iron-dependent lipid peroxidation. As a conservative evolution, ferroptosis exerts a critical role in healthy development or disease progression of various organisms (Distéfano et al. Citation2017). It is characterized by mitochondrial shrinkage and density increase morphologically and iron accumulation and lipid peroxidation biochemically (Xie et al. Citation2016).
Glucocorticoids are steroid hormones responding to environmental and physiological stimuli and can regulate the development and metabolism of various organisms extensively (Revollo and Cidlowski Citation2009). Glucocorticoids are widely applied to treat inflammatory and autoimmune diseases due to their potent anti-inflammatory and immunosuppressive activities. However, long-term or high-dose administration of dexamethasone (DEX, a glucocorticoid) can cause several side effects, such as secondary osteoporosis, osteonecrosis, skeletal muscle weakness, and cardiovascular disease (Ramamoorthy and Cidlowski Citation2013), which may be correlated with elevated autophagy and involvement of ferroptosis (Yang et al. Citation2021). Autophagy, an intracellular degradation system, is closely correlated with ferroptosis. Excessive autophagy can promote ferroptosis through iron accumulation or lipid peroxidation (Lee et al. Citation2020).
In summary, ferroptosis may be associated with the DEX-induced side effects, in which autophagy may be involved. However, the specific mechanism of ferroptosis in DEX-induced side effects remains unclear. Further understanding on the effects of ferroptosis in DEX-induced side effects is helpful in treating many diseases. This review summarizes the molecular mechanisms of ferroptosis, the DEX-induced side effects, and the potential relationship between them.
The mechanism of ferroptosis
Ferroptosis is caused by iron accumulation and lipid peroxidation. Meanwhile, ferroptosis may be regulated by various molecules and signals involved in iron metabolism, lipid peroxidation, and antioxidant defense.
Iron metabolism
Iron uptake
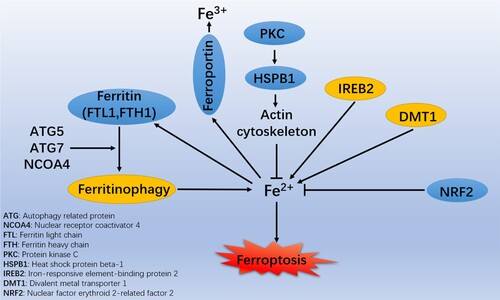
The control of cellular iron levels is dynamic. Iron usually is found as Fe2+ and Fe3+. Fe3+ enters cells through the transferrin / transferrin receptor (TF / TFRC) and is then reduced to Fe2+ by six-transmembrane epithelial antigen of prostate 3 (STEAP3). Finally, divalent metal transporter 1 / solute carrier family 11 member 2 (DMT1/SLC11A2) mediates the release of Fe2+ from the endosome into the labile iron pool (LIP). Under iron overload, excess iron may overwhelm the carrying capacity of TFRC and circulate in the form of non-TF-bound iron (NTBI) and then import Fe2+ through transmembrane transporters (Knutson Citation2019). In cells, heme is processed by cytosolic heme oxygenase 1 (HMOX1), releasing Fe2+ (Imoto et al. Citation2018). Excess iron can generate reactive oxygen species (ROS) through the iron-dependent Fenton reaction and the activation of iron-containing enzymes to promote lipid peroxidation, with excessive lipid peroxide deposition promoting ferroptosis (Li et al. Citation2012). However, HMOX1 is anti-ferroptotic in hepatocellular carcinoma cells due to its antioxidant activity (Sun et al. Citation2016). The roles of HMOX1 in ferroptosis are tightly related to its expression and the cell type. Protein kinase C (PKC) enhances the phosphorylation of heat shock protein beta-1 (HSPB1) and facilitates ferroptotic resistance by restricting the cytoskeleton-mediated iron absorption and subsequent ROS generation (Sun et al. Citation2015). In addition, the development of ferroptosis can be affected by iron transport among cytosol, mitochondria, and lysosomes, and activation of iron-responsive element-binding protein (IREB2) (Dixon et al. Citation2012) (Figure ).
Figure 1. Roles of iron metabolism in the molecular mechanisms and signaling pathways of ferroptosis.
Iron utilization
Generally, most iron in the LIP can be transferred to mitochondria to synthesize heme or iron-sulfur (Fe-S) clusters to protect cancer cells from ferroptosis (Alvarez et al. Citation2017). Poly(RC)-binding protein 1 (PCBP1) and PCBP2 can regulate the metalation of iron-containing proteins and the storage and export of Fe2+ (Philpott and Jadhav Citation2019). In the cytoplasm, iron can be delivered by hypoxia-inducible factor (HIF) prolyl hydroxylases through PCBP1 and PCBP2 (Nandal et al. Citation2011). PCBP1 can inhibit the toxicity of cytosolic iron and ferroptosis (Protchenko et al. Citation2021). Intracellular iron chelators such as deferoxamine (DFO) can block ferroptosis while erastin can induce ferroptosis. Iron chelators can significantly inhibit the cell death triggered by erastin (e.g. DFO), which means that iron-dependent signaling is conductive to the erastin-induced ferroptosis.
Iron storage
Excess iron is stored as ferritin, and an iron storage protein complex contains ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1). The ferritin can be degraded by nuclear receptor coactivator 4 (NCOA4) – mediated ferritinophagy to release iron (Gao et al. Citation2016; Hou et al. Citation2016). In addition to selective autophagy, ferritin can be degraded by activating the ubiquitin-proteasome system (UPS) (De Domenico et al. Citation2006). Thus, UPS and autophagy can maintain the intracellular ferritin levels. In the study of neuronal cells, Wang et al. (Citation2016) found that the overexpressed mitochondrial ferritin, which is an iron-storage protein, can diminish ferroptosis. Nuclear factor erythroid 2-related factor 2 (NRF2) can exert an anti-ferroptotic role, which can be reflected in promoting the transcription of genes encoding antioxidant proteins (such as heme oxygenase-1 [HO-1]) and iron metabolism proteins (such as FTH1), thereby inhibiting ferroptosis (Sun et al. Citation2016). These results indicate that iron-storage proteins can play a broad anti-ferroptotic role (Figure ).
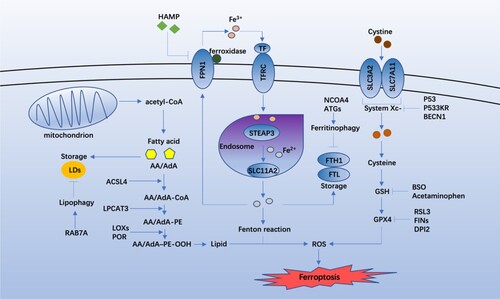
Figure 2. Core regulators of the molecular mechanisms and signaling pathways of ferroptosis.
Iron export
The iron-efflux protein solute carrier family 40 member 1 (SLC40A1/ferroportin/FPN) can transport excess Fe2+ out of cells, during which the Fe2+ is reoxidized to Fe3+ (Ma et al. Citation2016). SLC40A1 is negatively regulated by hepcidin antimicrobial peptide (HAMP), and the SLC40A1 is degraded in lysosomes after interaction with HAMP, reducing iron export and increasing susceptibility to ferroptosis. Mutations in the SLC40A1 gene can damage the SLC40A1 transport activity and result in iron overload-mediated ferroportin-related diseases (Détivaud et al. Citation2013) (Figure ).
Antioxidant defence
System Xc- is an amino acid antiporter formed by functional subunit solute carrier family 7 member 11 (SLC7A11) and the regulatory subunit SLC3A2. It is an essential antioxidant system in cells and maintains the production of glutathione (GSH) by exchanging extracellular cystine with intracellular glutamate (Bridges et al. Citation2012) during which the cystine, a precursor for GSH synthesis, is reduced to cysteine (Lewerenz et al. Citation2013; Sato et al. Citation1999). Small molecule compounds or drugs can inhibit SLC7A11, leading to GSH depletion and ferroptosis (Dixon et al. Citation2014). The p53 protein can inhibit the SLC7A11 (a key component of system Xc-) directly, restricting the cellular uptake of cysteine (Jiang et al. Citation2015). An acetylation-defective mutant of p53, p533KR, can’t cause cell cycle arrest, apoptosis, or senescence, but can induce ferroptosis by inhibiting SLC7A11 (Jiang et al. Citation2015). BECN1, a core player in autophagy regulation, can block system Xc- by binding to SLC7A11, thereby promoting the ferroptosis (Song et al. Citation2018).
GSH is a necessary cofactor of the glutathione peroxidase 4 (GPX4) antioxidant response, and the targeted regulation of GSH is an essential mechanism of ferroptosis (Forcina and Dixon Citation2019). Buthioninesulfoximine (BSO), an irreversible inhibitor of cysteine synthetase, can inhibit the synthesis of cysteine and GSH and eventually reduce the GPX4 activity and ferroptosis (Yang et al. Citation2014). In addition, the reactive metabolite of acetaminophen, N-acetyl-p-benzoquinone imine, can lead to GSH depletion and ferroptosis (Lőrincz et al. Citation2015).
GPX4 can reduce the toxic peroxides and exert a critical role in preventing the accumulation of ROS and maintaining the lipid homeostasis, ultimately blocking ferroptosis (Maiorino et al. Citation2018). Renin-angiotensin system (RAS) selective lethal 3 (RSL3) can inhibit GPX4 and induce ferroptosis, which irreversibly inhibits the activity of GPX4 by covalently targeting selenocysteine (Yang et al. Citation2014; Yang et al. Citation2016; Ingold et al. Citation2018). Ferroptosis-inducing agents (FINs) are inducers of ferroptosis (Yang et al. Citation2012; Weïwer et al. Citation2012). FINs in Class I, such as diverse pharmacological inhibitor 2 (DPI2), inhibit GPX4 through GSH depletion, which is exactly same with the mechanism of BSO, while those in Class II inhibit the activity of GPX4 directly (Yang et al. Citation2014) (Figure ).
Lipid peroxidation
Polyunsaturated fatty acids (PUFAs) contain unstable carbon–carbon double bonds that are prone to lipid peroxidation and are necessary for ferroptosis (Yang et al. Citation2016). As one of PUFAs, arachidonic acid or adrenic acid (AA/AdA) is the primary substrate of lipid peroxidation in ferroptosis (Kagan et al. Citation2017). Acyl-CoA synthase 4 (ACSL4) and lysophospholipid acyltransferase 3 (LPCAT3) are essential in synthesis and remodeling of PUFAs (Dixon et al. Citation2015). ACSL4 catalyzes the combination of free AA/AdA and CoA, forming derivatives AA/AdA–CoA, and LPCAT3 then promotes their esterification to membrane phosphatidylethanolamine (PE) to form AA/AdA–PE. Different lipoxygenases with various contexts can mediate lipid peroxidation to produce the hydroperoxides AA/AdA–PE-OOH, promoting ferroptosis (Figure ). When these two genes are knocked out, the synthesis of PUFAs weakens, inhibiting ferroptosis (Kagan et al. Citation2017; Doll et al. Citation2017). Therefore, PUFAs are essential targets for plasma membrane peroxidation during ferroptosis.
The non-enzymatic lipid peroxidation results from an attack on polyunsaturated lipids by hydroxyl radicals (HO•) generated in the Fenton reaction Fe2+ + H2O2 → Fe3+ + HO• + HO (Koppenol Citation2001; Lai and Piette Citation1978). Enzymatic lipid peroxidation is mediated by activity of lipoxygenases (LOXs). LOXs are lipid metabolizing enzymes to essentially mediate the PUFA peroxidation to produce AA/AdA-PE-OOHs, thus causing ferroptosis (Kuhn et al. Citation2015). Cytochrome P450 oxidoreductase (POR) can combine with flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) to provide electrons to the P450 enzyme, thereby promoting PUFA peroxidation in a LOX-independent manner (Zou et al. Citation2020) (Figure ).
Ferroptosis inhibitors
Most specific ferroptosis inhibitors are antioxidants and iron chelators. Antioxidants, including ferro-statin-1 (Fer-1), lipoxstatin-1, zileuton, and vitamin E, hinder ferroptosis by inhibiting the formation of lipid peroxides (Li et al. Citation2020).
Ferroptosis and inflammation
Ferroptosis plays a crucial role in inflammation. In some disease models, several antioxidants with ferroptosis inhibitors have shown anti-inflammatory effects. During the cell stress, AA is released from phospholipids and metabolized into inflammatory mediators through cyclooxygenase (COX) and LOX (Wang et al. Citation2019). Ferroptosis can promote the activity of COX and LOX by releasing oxidized lipid mediators (Yang et al. Citation2014), and ferroptotic cells can be undertaken as donors of AA for the transcellular biosynthesis of eicosanoids (Folco and Murphy Citation2006). In addition, ferroptosis triggers inflammation by releasing damage-associated molecular patterns (DAMPs), which are immunogenic (Kim et al. Citation2019). Some inflammatory cytokines interact between ferroptosis and inflammation (Sun et al. Citation2020). Ferroptosis inhibitors are of significant benefit in certain diseases through their anti-inflammatory effects (Sun et al. Citation2020). However, ferroptosis can protect the skin from inflammation (Arbiser et al. Citation2018). Therefore, ferroptosis may be a double-edged sword.
DEX-induced side effects
DEX shows potent anti-inflammatory and immunosuppressive activities but can cause many side effects. Programmed cell death pathways such as pyroptosis, necroptosis, and ferroptosis may play essential roles in the DEX-induced side effects.
Pyroptosis
Pyroptosis refers to a pro-inflammatory programmed cell death (Kroemer et al. Citation2005). Nucleotide oligomerization domain (NOD)-like receptors (NLRs) are upstream activators of pyroptosis with the best characterization effect and are inflammasome-signaling pathway components (Xue et al. Citation2019). Activation of NLRP3 can trigger caspase-1-dependent pyroptosis (Ding et al. Citation2019). Skeletal muscle atrophy is a significant side effect caused by high doses or sustained usage of glucocorticoids (Schakman et al. Citation2013). Pyroptosis plays an important role in the development of muscle atrophy (Ding et al. Citation2019). Wang et al. (Citation2021) found that DEX could induce muscle atrophy and pyroptosis. Knockdown of NLRP3 attenuates the DEX-induced muscle pyroptosis and atrophy, indicating that DEX activates the NLRP3 inflammasome to trigger myotube pyroptosis. Such result suggests that NLRP3 can be a therapeutic target of skeletal muscle atrophy.
Necroptosis
Necroptosis is a programmed cell necrosis based on the formation of necrotizing bodies instead of activation of caspases (Holler et al. Citation2000). Receptor interacting protein (RIP)−1 and RIP-3 are activated to form the necrosome complex, and necroptosis results in elevated ROS and decreased mitochondria membrane potential (MMP) (Shi et al. Citation2017). Feng et al. (Citation2019) found that necroptosis and apoptosis are essential to study the DEX-induced osteoporosis, and that treatment with necrostatin-1 and the specific inhibitor of necroptosis could significantly inhibit the DEX-induced necrotic-like cell death. The mechanism of necrostatin-1 protection against glucocorticoid-induced cell death may be mediated by caspase-independent pathways involving RIP-1 and RIP-3, ultimately reducing the MMP.
Ferroptosis
Ferroptosis is primarily caused by excessive accumulation of iron-dependent ROS in cells, weakening of the scavenging effect of GPX4 and homeostatic imbalances between production and degradation of ROS (Xie et al. Citation2016). Lu et al. (Citation2019) found that high doses of DEX can down-regulate the GPX4 and system Xc-. Gene enrichment analysis based on the Kyoto Gene and Genome Encyclopedia showed that the ferroptotic pathway is activated. Recently, Yang et al. (Citation2021) revealed that inhibiting osteoblast ferroptosis can reverse the DEX-induced osteoporosis. Such findings suggest that ferroptosis is involved in the DEX-induced side effects.
DEX and autophagy
Some studies on DEX-induced side effects pointed out that DEX can promote autophagy. In a mouse model of glucocorticoid-induced obesity, Deng et al. (Citation2020) revealed that DEX could enable brown adipose tissue (BAT) to acquire white adipose tissue (WAT) cell features, namely BAT whitening; autophagy and autophagy-related 7 (ATG7) expressions were induced in BAT by DEX; and autophagy inhibitor chloroquine or adenovirus-mediated ATG7 knockdown for treatment prevented the DEX-induced BAT whitening and fat mass gain. DEX is applied to induce maturation and improve the viability of preterm infants (Roberts and Dalziel Citation2006). However, it can reduce the placental and fetal weight in humans (Khan et al. Citation2011). Deng et al. (Citation2020) found that DEX downregulates SLC7A5 (the amino acid carrier) via GR-mediated transrepression. Uptake of impaired amino acid inhibits the mTOR signaling, causing inhibited proliferation and enhancing autophagy and apoptosis. Yang et al. (Citation2021) studied the DEX-induced osteoporosis and found that DEX promotes the expression change of the autophagy-related proteins, suggesting upregulated LC3 II, BECN-1, and NCOA4. Interestingly, ferroptosis is inhibited at suppressed autophagy, whereas ferroptosis is activated at DEX-induced autophagy (Yang et al. Citation2021).
Role of autophagy in ferroptosis
Autophagy is increasingly proved to be tightly associated with ferroptosis. Excessive autophagy can cause iron accumulation or lipid peroxidation, resulting in ferroptosis. Here, the key types of autophagy and the regulators of the autophagic machinery in driving ferroptosis are summarized as follows.
NCOA4-dependent ferritinophagy
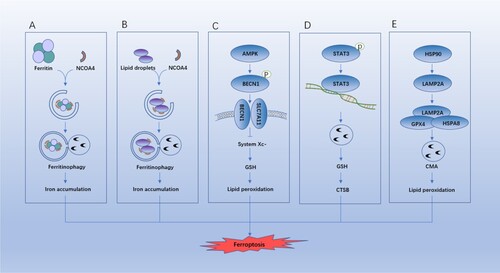
As an iron storage protein complex, ferritin can adjust the bioavailability of intracellular iron. Ferritinophagy refers to autophagic degradation of ferritin. Ferritinophagy can promote ferroptosis by degrading ferritin and elevate the iron level, causing oxidative injury by the Fenton reaction (Xie et al. Citation2016; Hou et al. Citation2016) (Figure (A)). NCOA4 is a selective cargo receptor that mediates ferritinophagy (Mancias et al. Citation2014). The knockdown or inhibition of NCOA4 or ATG proteins can play the roles of inhibiting ferritin degradation, reducing free iron levels, and limiting lipid peroxidation during ferroptosis (Xie et al. Citation2016; Hou et al. Citation2016) (Figure (A)).
Figure 3. Role of autophagy in ferroptosis. (A) NCOA4-mediated ferritinophagy promotes iron accumulation. (B) RAB7A-mediated lipophagy promotes lipid peroxidation. (C) BECN1-mediated system Xc- inhibition promotes GSH depletion and ferroptosis. (D) STAT3-mediated CTSB expression and release announce lysosomal cell death. (E) HSP90-mediated LAMP2A stability contributes to CMA-mediated GPX4 degradation.
RAB7A-dependent lipophagy
Lipid droplets (LDs) act as inert sites for fat fixation and change the storage and hydrolysis of neutral lipids in organelles (Liu and Czaja Citation2013). As a selective autophagy, lipophagy can lead to autophagic degradation of intracellular LDs and regulate the level of cellular lipids. RAB7A can mediate the recruitment of LDs during lipid phagocytosis. Bai et al. (Citation2019) pointed out that LDs are associated with oxidative stress-induced ferroptosis in a negative way; and RAB7A-dependent lipophagy can promote degradation of LDs and generation of free fatty acids, thus accelerating the lipid peroxidation-mediated ferroptosis (Schroeder et al. Citation2015) (Figure (B)).
BECN1-dependent system Xc- inhibition
BECN1 is a binding protein that regulates cellular processes such as autophagy and apoptosis, and its multifunctional role depends on its binding partners (Kang et al. Citation2011). Song et al. found that BECN1 is a SLC7A11 binding protein that contributes to cell death in response to type 1 ferroptosis inducers (e.g. erastin and sorafenib), which is controlled by PRKAA/AMPKα (adenosine monophosphate activated protein kinase alpha)-mediated phosphorylation of BECN1 at Ser90/93/96 (Song et al. Citation2018) (Figure (C)). Therefore, the mutated BECN1 phosphorylation sites or inhibited AMPK activity can inhibit ferroptosis by restricting the generation of BECN1-SLC7A11 complex.
STAT3-dependent lysosomal membrane permeabilization (LMP)
Lysosomal dysfunction is jointly caused by various regulatory cell deaths, including autophagy-dependent cell death (Kroemer and Jäättelä Citation2005). Lysosomes are conductive to promoting ferroptosis (Gao et al. Citation2018; Torii et al. Citation2016). Gao et al. (Citation2018) disclosed that signal transducer and activator of transcription 3 (STAT3) can positively regulate the ferroptosis, and STAT3 promotes the erastin-induced ferroptosis by inducing LMP and subsequent lysosomal cell death. Mechanically, STAT3 mediates the expression of CTSB (cathepsin B), causing ferroptosis. The lysosome-dependent cell death inhibited by pharmacological blockade of cathepsin activity (using CA-074Me) can restrict the erastin-induced ferroptosis (Gao et al. Citation2018) (Figure (D)).
HSP90-mediated chaperone-mediated autophagy (CMA)
CMA is a selective autophagy delivering specific proteins to lysosomes for degradation. CMA-induced GPX4 degradation was found in ferroptosis caused by various ferroptosis activators. Heat shock protein 90 (HSP90) is a major molecular chaperone regulating the apoptosis and necrosis (Jacobsen et al. Citation2016; Li et al. Citation2015; Zhao et al. Citation2016). Recently, HSP90 is proved to be an important element in ferroptosis, which is reflected in increasing the protein stability of lysosomal-associated membrane protein 2A (LAMP2A) to promote the GPX4 degradation mechanistically (Wu et al. Citation2019) (Figure (E)).
Potential relationship between ferroptosis and DEX-induced side effects
In conclusion, this work shown that autophagy and ferroptosis are involved in DEX-induced side effects, and autophagy can promote the occurrence of ferroptosis. Therefore, DEX-induced side effects may be related to DEX, autophagy, and ferroptosis. In addition, some possible ways were proposed to explain the relationships mentioned.
BECN1-mediated inhibition of system Xc-
The above review on the role of autophagy in ferroptosis revealed that adenosine monophosphate activated protein kinase (AMPK)-mediated BECN1 phosphorylation promotes the formation of the BECN1-SLC7A11 complex and the inhibition of system Xc-, thereby reducing antioxidants (GPX4 and GSH) and causing ferroptosis. The study on DEX-induced osteoporosis found that ferroptosis is involved. DEX could promote autophagy (upregulated BECN1), and ferroptosis was inhibited at suppressed autophagy, whereas ferroptosis was activated at DEX-induced autophagy. Therefore, it was speculated the pathway that occurred in DEX-induced osteoporosis was reflected in DEX-increased BECN1. In addition, the formation of the BECN1-SLC7A11 complex and inhibition of system Xc− were simulated in this work, and the results suggested that the antioxidants (GPX4 and GSH) were reduced, leading to ferroptosis. However, in DEX-increased BECN1, whether AMPK played a regulatory role in mechanism has not been studied.
Autophagy-related protein (ATG) 5 /ATG7/nuclear receptor coactivator 4 (NCOA4) mediated ferritinophagy
NCOA4 or ATGs can promote the degradation of ferritin, iron accumulation, lipid peroxidation, and subsequent ferroptosis. The study of DEX-induced osteoporosis found that ferroptosis was involved. DEX could promote autophagy (upregulated NCOA4). The expression of NCOA4 transported ferritin to the autophagosome. Therefore, it was speculated that the DEX promoted autophagy through ATG5/ATG7/NCOA4 signaling and caused ferritin degradation, iron accumulation, and lipid accumulation peroxidation, ultimately promoting ferroptosis and resulting in a series of side effects.
Conclusions
Long-term or high-dose administration of DEX can cause some severe side effects. Ferroptosis is increasingly proved to play an important effect in many diseases. However, there are few studies on whether DEX-induced side effects are related to ferroptosis. A deeper understanding on the role of ferroptosis in DEX-induced side effects will help in treating many diseases. Here, some possible ways to explain the relationship between ferroptosis and DEX-induced side effects are proposed. Interestingly, ferroptosis could mediate the inflammatory response, so it is suspected that inhibiting ferroptosis could reduce DEX-induced side effects and enhance its anti-inflammatory effect. However, this conjecture has to be further verified.
Acknowledgements
HongJiang Zhao and HongKai Shang contributed to the conception or design of the work. HongJiang Zhao drafted the manuscript. HongJiang Zhao and HongKai Shang revised the manuscript for important intellectual content and approved the final manuscript. Both authors agree to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K, Possemato R. 2017. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 551(7682):639–643.
- Arbiser JL, Bonner MY, Ward N, Elsey J, Rao S. 2018. Selenium unmasks protective iron armor: a possible defense against cutaneous inflammation and cancer. Biochim Biophys Acta Gen Subj. 1862(11):2518–2527.
- Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, Kang R, Wang X, Tang D, Dai E. 2019. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 508(4):997–1003.
- Bridges RJ, Natale NR, Patel SA. 2012. System xc(-) cystine/ glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 165(1):20–34.
- De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J. 2006. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. Embo J. 25(22):5396–5404.
- Deng J, Guo Y, Yuan F, Chen S, Yin H, Jiang X, Jiao F, Wang F, Ji H, Hu G, et al. 2020. Autophagy inhibition prevents glucocorticoid-increased adiposity via suppressing BAT whitening. Autophagy. 16(3):451–465.
- Détivaud L, Island ML, Jouanolle AM, Ropert M, Bardou-Jacquet E, Le Lan C, Mosser A, Leroyer P, Deugnier Y, David V, et al. 2013. Ferroportin diseases: functional studies, a link between genetic and clinical phenotype. Hum Mutat. 34(11):1529–1536.
- Ding J, Li F, Cong Y, Miao J, Wu D, Liu B, Wang L. 2019. Trichostatin A inhibits skeletal muscle atrophy induced by cigarette smoke exposure in mice. Life Sci. 235:116800.
- Distéfano AM, Martin MV, Córdoba JP, Bellido AM, D'Ippólito S, Colman SL, Soto D, Roldán JA, Bartoli CG, Zabaleta EJ, et al. 2017. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 216(2):463–476.
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149(5):1060–1072.
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, et al. 2014. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 3:e02523.
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR. 2015. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 10(7):1604–1609.
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. 2017. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 13(1):91–98.
- Feng M, Zhang R, Yang P, Wang K, Qiang H. 2019. Interaction between necroptosis and apoptosis in MC3T3-E1 cell death induced by dexamethasone. J South Med Univ. 39(9):1030–1037.
- Folco G, Murphy RC. 2006. Eicosanoid transcellular biosynthesis: from cell-cell interactions to in vivo tissue responses. Pharmacol Rev. 58(3):375–388.
- Forcina GC, Dixon SJ. 2019. Gpx4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 19(18):e1800311.
- Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, Dai E. 2018. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun. 503(3):1550–1556.
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. 2016. Ferroptosis is an autophagic cell death process. Cell Res. 26(9):1021–1032.
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 1(6):489–495.
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, 3rd, Kang R, Tang D, 2016. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 12(8):1425–1428.
- Imoto S, Kono M, Suzuki T, Shibuya Y, Sawamura T, Mizokoshi Y, Sawada H, Ohbuchi A, Saigo K. 2018. Haemin-induced cell death in human monocytic cells is consistent with ferroptosis. Transfus Apher Sci. 57(4):524–531.
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. 2018. Selenium utilization by GPX4 Is required to prevent hydroperoxide-induced ferroptosis. Cell. 172(3):409–422.
- Jacobsen AV, Lowes KN, Tanzer MC, Lucet IS, Hildebrand JM, Petrie EJ, van Delft MF, Liu Z, Conos SA, Zhang JG, et al. 2016. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 7(1):e2051.
- Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, Baer R, Gu W. 2015. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 520(7545):57–62.
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. 2017. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 13(1):81–90.
- Kang R, Zeh HJ, Lotze MT, Tang D. 2011. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18(4):571–580.
- Khan AA, Rodriguez A, Kaakinen M, Pouta A, Hartikainen AL, Jarvelin MR. 2011. Does in utero exposure to synthetic glucocorticoids influence birthweight, head circumference and birth length? A systematic review of current evidence in humans. Paediatr Perinat Epidemiol. 25(1):20–36.
- Kim EH, Wong SW, Martinez J. 2019. Programmed necrosis and disease: we interrupt your regular programming to bring you necroinflammation. Cell Death Differ. 26(1):25–40.
- Knutson MD. 2019. Non-transferrin-bound iron transporters. Free Radic Biol Med. 133:101–111.
- Koppenol WH. 2001. The haber-weiss cycle–70 years later. Redox Rep. 6(4):229–234.
- Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight, et al. 2005. Classification of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ. 12(Suppl 2):1463–1467.
- Kroemer G, Jäättelä M. 2005. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 5(11):886–897.
- Kuhn H, Banthiya S, van Leyen K. 2015. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta. 1851(4):308–330.
- Lai CS, Piette LH. 1978. Spin-trapping studies of hydroxyl radical production involved in lipid peroxidation. Arch Biochem Biophys. 190(1):27–38.
- Lee YS, Kalimuthu K, Seok Park Y, Makala H, Watkins SC, Choudry MHA, Bartlett DL, Tae Kwon Y, Lee YJ. 2020. Ferroptotic agent-induced endoplasmic reticulum stress response plays a pivotal role in the autophagic process outcome. J Cell Physiol. 235(10):6767–6778.
- Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, et al. 2013. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 18(5):522–555.
- Li D, Xu T, Cao Y, Wang H, Li L, Chen S, Wang X, Shen Z. 2015. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci U S A. 112(16):5017–5022.
- Li GF, Pan YZ, Sirois P, Li K, Xu YJ. 2012. Iron homeostasis in osteoporosis and its clinical implications. Osteoporos Int. 23(10):2403–2408.
- Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang G. 2020. Ferroptosis: past, present and future. Cell Death Dis. 11(2):88.
- Liu K, Czaja MJ. 2013. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 20(1):3–11.
- Lőrincz T, Jemnitz K, Kardon T, Mandl J, Szarka A. 2015. Ferroptosis is involved in Acetaminophen induced cell death. Pathol Oncol Res. 21(4):1115–1121.
- Lu J, Yang J, Zheng Y, Chen X, Fang S. 2019. Extracellular vesicles from endothelial progenitor cells prevent steroid-induced osteoporosis by suppressing the ferroptotic pathway in mouse osteoblasts based on bioinformatics evidence. Sci Rep. 9(1):16130.
- Ma S, Henson ES, Chen Y, Gibson SB. 2016. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 7(7):e2307.
- Maiorino M, Conrad M, Ursini F. 2018. Gpx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signaling. 29(1):61–74.
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. 2014. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 509(7498):105–109.
- Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, Bruick RK, Philpott CC. 2011. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 14(5):647–657.
- Philpott CC, Jadhav S. 2019. The ins and outs of iron: escorting iron through the mammalian cytosol. Free Radic Biol Med. 133:112–117.
- Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, Ghosh MC, Cox JE, Maschek JA, Tyurin VA, et al. 2021. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology. 73(3):1176–1193.
- Ramamoorthy S, Cidlowski JA. 2013. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr Dev. 24:41–56.
- Revollo JR, Cidlowski JA. 2009. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 1179:167–178.
- Roberts D, Dalziel S. 2006. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 3:Cd004454.
- Sato H, Tamba M, Ishii T, Bannai S. 1999. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 274(17):11455–11458.
- Schakman O, Kalista S, Barbé C, Loumaye A, Thissen JP. 2013. Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol. 45(10):2163–2172.
- Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. 2015. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 61(6):1896–1907.
- Shi J, Gao W, Shao F. 2017. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 42(4):245–254.
- Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al. 2018. AMPK-Mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X activity. Curr Biol. 28(15):2388–2399.
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. 2016. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 63(1):173–184.
- Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, Wang H, Cao L, Tang D. 2015. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 34(45):5617–5625.
- Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, Xu S, Gao Y, Chen X, Sui X, et al. 2020. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. 127:110108.
- Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, et al. 2016. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 473(6):769–777.
- Wang L, Jiao XF, Wu C, Li XQ, Sun HX, Shen XY, Zhang KZ, Zhao C, Liu L, Wang M, et al. 2021. Trimetazidine attenuates dexamethasone-induced muscle atrophy via inhibiting NLRP3/GSDMD pathway-mediated pyroptosis. Cell Death Discov. 7(1):251.
- Wang T, Fu X, Chen Q, Patra JK, Wang D, Wang Z, Gai Z. 2019. Arachidonic acid metabolism and kidney inflammation. Int J Mol Sci. 20(15):3683.
- Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, Yu P, Shi ZH, Wu WS, Gao G, et al. 2016. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. 8:308.
- Weïwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber, et al. 2012. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett. 22(4):1822–1826.
- Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, Shan B, Pan H, Yuan J. 2019. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 116(8):2996–3005.
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. 2016. Ferroptosis: process and function. Cell Death Differ. 23(3):369–379.
- Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. 2019. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 40(11):1035–1052.
- Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B, Jiang LS, Jiang SD. 2021. Exosomes derived from vascular endothelial cells antagonize glucocorticoid-induced osteoporosis by inhibiting ferritinophagy with resultant limited ferroptosis of osteoblasts. J Cell Physiol. 236(9):6691–6705.
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. 2016. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 113(34):E4966–E4E75.
- Yang WS, Shimada K, Delva D, Patel M, Ode E, Skouta R, Stockwell BR. 2012. Identification of simple compounds with microtubule-binding activity that inhibit cancer cell growth with high potency. ACS Med Chem Lett. 3(1):35–38.
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. 2014. Regulation of ferroptotic cancer cell death by GPX4. Cell. 156(1–2):317–331.
- Zhao XM, Chen Z, Zhao JB, Zhang PP, Pu YF, Jiang SH, Hou JJ, Cui YM, Jia XL, Zhang SQ. 2016. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 7(2):e2089.
- Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, Schreiber SL. 2020. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 16(3):302–309.