
Abstract
The central nervous system (CNS) can be preconditioned to resist damage by peripheral pretreatment with low-dose gram-negative bacterial endotoxin lipopolysaccharide (LPS). Underlying mechanisms associated with transient protection of the cerebral cortex against traumatic brain injury include increased neuronal production of antiapoptotic and neurotrophic molecules, microglial-mediated displacement of inhibitory presynaptic terminals innervating the soma of cortical projection neurons, and synchronized firing of cortical projection neurons. However, the cell types and signaling responsible for these neuronal and microglial changes are unknown. A fundamental question is whether LPS penetrates the CNS or acts on the luminal surface of brain endothelial cells, thereby triggering an indirect parenchymal neuroprotective response. The present study shows that a low-dose intraperitoneal LPS treatment increases brain endothelial cell activation markers CD54, but does not open the blood–brain barrier or alter brain endothelial cell tight junctions as assessed by electron microscopy. NanoString nCounter transcript analyses of CD31-positive brain endothelial cells further revealed significant upregulation of Cxcl10, C3, Ccl2, Il1β, Cxcl2, and Cxcl1, consistent with identification of myeloid differentiation primary response 88 (MyD88) as a regulator of these transcripts by pathway analysis. Conditional genetic endothelial cell gene ablation approaches demonstrated that both MyD88-dependent Toll-like receptor 4 (TLR4) signaling and Cxcl10 expression are essential for LPS-induced neuroprotection and microglial activation. These results suggest that C-X-C motif chemokine ligand 10 (CXCL10) production by endothelial cells in response to circulating TLR ligands may directly or indirectly signal to CXCR3 on neurons and/or microglia. Targeted activation of brain endothelial receptors may thus provide an attractive approach for inducing transient neuroprotection.
Introduction
Development of neuroprotective therapies for individuals with acute or chronic brain diseases is a priority in neuroscience research. Neuroprotective approaches include increased expression of neurotrophic factors, reduction of reactive oxygen species, and inhibition of neuronal apoptosis (CitationChen and Trapp, 2016; CitationFaden and Stoica, 2007). However, targeting these neuroprotective mediators requires penetration of therapeutic agents into the central nervous system (CNS). The blood–brain barrier (BBB) presents a formable obstacle to the development of therapies that are designed to mitigate neurodegeneration in this manner (CitationCho et al., 2017). Therefore, neuroprotective strategies that circumvent brain penetration would constitute a major advance.
A well-established paradigm of prophylactic neuroprotection in rodents is based on preconditioning of the CNS via peripherally administered low-dose endotoxin lipopolysaccharide (LPS; for review, see CitationSangaran et al., 2021). The low-dose intraperitoneal (IP) LPS treatment paradigm activates microglia and protects against traumatic brain injury (TBI). Dosage, frequency, and timing of LPS treatment are critical for effective prophylaxis, and the neuroprotection afforded by four daily LPS IP injections is insult specific. A single injection of LPS transiently protects against ischemic infarcts, but does not activate microglia nor protect against cryogenic-induced cortical injury (Stevens et al., Citation2011). By contrast, four IP injections of LPS confer protection against TBI but not ischemic stroke. Thus, it appears that two LPS-induced neuroprotective pathways can be induced: one protecting against ischemic brain injury and one against TBI. The former is likely to be mediated at the level of the CNS vasculature and involves interferon signaling (Stevens et al., 2011). Protection from TBI using daily injections of LPS for four consecutive days involves microglial activation and their displacement of inhibitory presynaptic terminals from cortical projection neurons (CitationChen et al., 2012). This displacement enhances synaptic N-methyl-D-aspartate receptor firing (CitationHardingham et al., 2001), activation of Ca2+/calmodulin-dependent kinase IV, phosphorylation of cAMP responses element binding protein (CREB), and neuronal expression of antiapoptotic and neurotrophic molecules, which results in reduced apoptosis of cortical neurons and lesion volume following TBI (CitationChen et al., 2014).
LPS binds to Toll-like receptor 4 (TLR4), which is expressed on monocytes (CitationChen et al., 2012), microglia (CitationVaure and Liu, 2014), and brain endothelial cells (CitationNagyoszi et al., 2010; CitationVanlandewijck et al., 2018); its expression on murine astrocytes has not been established (CitationVaure and Liu, 2014). Reciprocal bone-marrow chimeras between wild-type (WT) and TLR4-null mice established that TLR4 expression by tissue-resident cells such as endothelial and/or radiation-resistant microglia, but not by bone marrow-derived immune cells (e.g., monocytes) was essential for LPS-induced neuroprotection (CitationChen et al., 2012).
The present study investigated whether low-dose IP-injected LPS for four days activates TLR4 on the luminal surface of brain blood vessels or enters the brain and activates TLR4 on microglia. Electron microscopy (EM) and analysis of tight junction (TJ) proteins revealed that LPS directly activates brain endothelial cells without altering the BBB. Moreover, LPS induced an endothelial cell cytokine storm that included high levels of cxcl10 messenger RNA (mRNA). Using conditional endothelial cell-specific gene ablation, both myeloid differentiation primary response 88 (MyD88)-dependent TLR4 signaling and CXCL10 expression were found to be essential for LPS-induced microglial activation and neuroprotection. These studies support the novel concept that transient neuroprotection can be induced by directly targeting the activation of vascular endothelial cells, thereby circumventing the need for neuroprotective drug penetration into the brain parenchyma.
Materials and Methods
Animals
Only adult male mice were used in this study to be consistent with published studies investigating LPS-induced neuroprotection (CitationChen et al., 2014). C57BL/6J (B6) and Cxcl10−/− mice at 8 to 12 weeks of age were purchased from Jackson Labs. Myd88fl/fl and Tie2e-Cre mice were provided by Dr. Xiaoxia Li (Cleveland Clinic). CXCL10fl/fl mice were provided by Dr. David Pleasure (UC Davis). All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic.
LPS Treatment
Mice received one or four daily IP injections of LPS (Sigma Escherichia coli, serotype 055:B5) in phosphate-buffered saline (PBS) at 1.0 mg/kg. Control animals received similar injections of PBS. For lateral ventricle administration of purified murine CXCL10 (R&D systems) or anti-CXCL10 IgG (Cell Signaling), a catheter (Plastics One) was implanted into the brain (posterior: −0.5 mm; lateral: 1 mm; depth: 2.5 mm, relative to bregma) and fixed in place with dental acrylic 7 days before the delivery of the compounds. CXCL10 (in 5 μL artificial cerebrospinal fluid [CSF], Harvard Instruments) was delivered into the lateral ventricle daily for four consecutive days with a microsyringe (Hamilton) at a rate of 1 μL/min controlled by a microinjection pump (KD Scientific). Mice were sacrificed 24 hr after the final injection. For anti-CXCL10 IgG administration, 1 μg of antibody dissolved in artificial CSF was delivered at a rate of 1 μL/min 4 hr before the first and third injections of LPS.
Immunohistochemistry and Morphometric Analysis
Mice were anesthetized and transcardially perfused with freshly prepared 4% paraformaldehyde in 0.08 M phosphate buffer (pH = 7.4). Brains were removed, postfixed overnight, cryoprotected in 20% glycerol, and sectioned on a freezing sliding microtome. Free-floating coronal brain sections (30 µm) were pretreated with 3% H2O2, and 1% Triton X-100 in PBS and blocked with 3% normal goat serum. Sections were incubated in primary antibodies (Iba-1, generated at the Cleveland Clinic Hybridoma Core) overnight and labeled with the avidin-biotin complex (Vector Laboratories), and visualized with diaminobenzidine (DAB; Sigma). The DAB reaction product was enhanced with 0.04% osmium tetroxide (Electron Microscopy Sciences) for 30 s sections mounted on glass slides in glycerol and cover-slipped for microscopic analysis. Digital images were photographed using a Leica DMR microscope fitted with an Optronics Magna Fire CCD color video camera and analyzed with ImageJ software. On average, two to three sections per mouse were analyzed, and four micrographs at 20× were taken from each section. The average from all these regions of interest was computed to represent a single mouse. One symbol in plots thus represents 8 to 12 images per mouse.
Flow Cytometry Analysis of Brain Endothelial Cells
Brains were dissected and minced into small pieces (<1 mm3). Single-cell suspensions were generated by incubating the brain tissues with 0.5% Collagenase/Dispase (Roche) and 20 U DNase (Sigma) at 37°C for 75 min with occasional agitation. Cell suspensions were washed and centrifuged at 800g over a 30%/70% Percoll (GE Healthcare) gradient. Cells collected at the interphase were washed with FACS buffer (1% bovine serum albumin and 0.1% NaN3 in PBS) and blocked with FcR blocker (BD Pharmingen) on ice for 30 min. Fluorochrome-conjugated primary antibodies (FITC-CD31 [BD] or BV650-CD31 [BD] in combination with BV785-CD45 [BD], PE-TLR4 [BD], PE-CD54 [ThermoFisher], or APC-CD105 [ThermoFisher], all at 1:100 dilution), were added to the cells, which remained on ice for 30 min. Appropriate fluorochrome-conjugated isotype controls were used in parallel. Stained cells were acquired on a FACSCalibur (Becton Dickinson) or CytoFLEX LX (Beckman Coulter) flow cytometer and the resulting data were analyzed with Flowjo 10 software (Tree Star).
Immunofluorescence and Confocal Microscopy
For double or triple staining, free-floating coronal brain sections (30 µm) were pretreated with 1% Triton X-100 in PBS and blocked with 3% normal goat serum. Sections were incubated with primary antibodies (mouse-anti-Iba-1, 1:500, CCF Hybridoma Core; rat-anti-F4/80, 1:200, Abd Serotech; rabbit-anti-GAD65/67, 1:250, Millipore) overnight at 4°C and then stained with appropriate Alexa-488 or -647 conjugated secondary antibodies (Vector Laboratories) for 2 hr at room temperature. During the last 20 min of incubation, neurons were stained using Neuro Trace Nissl stain 530/615 (Molecular Probes) at 1:200. Sections were rinsed, mounted with a vectashield (Vector Laboratories), and examined in a Leica TCS confocal microscope (Leica Microsystems). The percentage of neuronal circumference covered by inhibitory synapses was calculated by dividing the length of glutamic acid decarboxylase (GAD) 65/67 puncta around the neuronal soma by neuronal circumference. Four mice were included in each group for this analysis. Ten to 15 neurons were analyzed from each mouse.
To assess blood vessel areas in close proximity to microglia processes, brain sections were stained with rat-anti-F4/80 (1:200, AbD Serotec) and rabbit-anti-CD31 (1:50, Abcam) as described above. Blood vessels were viewed in longitudinal orientation to capture long asymmetrically oriented microglia processes that line blood vessels. Specific lengths of blood vessels defined by arrows were analyzed for fluorescent intensities of anti-CD31 and F4/80 staining along a defined length.
BBB Permeability Assessment
To detect fluid-phase shifts between the circulation and CNS, BBB permeability was assessed using sodium-fluorescein (NaF). Briefly, mice received 100 μL of 10% NaF in PBS (IP) and cardiac blood was collected 10 min later. Mice were transcardially perfused with 10 mL of PBS and brains were removed. Each tissue was weighed, homogenized in PBS using a TissueLyser (Qiagen), and centrifuged at 14,000 g for 2 min. A volume of 500 μL of clarified supernatant was added to 500 μL of 15% trichloroacetic acid (Sigma) and centrifuged at 10,000 g for 10 min. A volume of 125 μL of 5 N NaOH (Sigma-Aldrich) was added to 500 μL of the supernatant and NaF content was measured on a SpectraMax Mz (Molecular Devices) microplate reader using standards ranging from 125 to 4,000 μg. NaF in the brain was normalized to sera NaF content using the formula: (μg of NaF in brain tissue per mg of protein)/(μg of NaF in sera per μL of blood). Data are expressed as fold increases in NaF in the brain, with levels from PBS-treated mice set at 1. For the positive control, C57BL/6 mice were infected at 6 to 7 weeks of age by intracranial injection into the left cerebral hemisphere with 1,000 plague-forming units (PFU) of the J.2.2v-1 monoclonal Ab-derived glia-tropic JHMV variant in 30 μL sterile PBS (CitationFleming et al., 1986; CitationPhares et al., 2014).
NanoString Analysis
Single-cell brain suspensions were obtained as described above. The cells were incubated with fluorescently conjugated CD31, CD11b, and CD45 antibodies (1:100, BD Pharmingen) at 4°C for 30 min. Fluorescently labeled cell populations were separated using a Becton-Dickinson FACSAria I Flow Sorter. The purity of the isolated endothelial cells (CD31+) was routinely over 90%, and that of microglia (CD11b+CD45lo) was over 95%. For NanoString analysis, total RNAs from purified endothelial cells were isolated using RNeasy Mini kits (Qiagen) and quantified with a NanoDrop spectrophotometer (Thermo). RNA from each sample (150 ng) was couriered on dry ice to NanoString Technologies in Seattle, WA. Mouse inflammation CodeSet was used to evaluate the differential expression of the genes of interest. Normalized data were electronically returned from NanoString for further in-house analysis. NanoString data were deposited at Gene Expression Omnibus and can be accessed using accession number GSE193015.
Real-Time Polymerase Chain Reaction and Cell-Specific Genotyping
For quantitative real-time polymerase chain reaction (RT-PCR), cDNA was synthesized by using the high-capacity RNA-to-cDNA kit (Applied Biosystems). RT-PCR was performed using TaqMan gene expression technology (Applied Biosystems) in duplicate on a 7300 RT-PCR system (Applied Biosystems). TaqMan probe/primer sets used in this study are a preassembled proprietary mix and are readily available from Life Technologies. PCR conditions were as follows: 10 min at 95°C, followed by 40 cycles at 95°C for 10 s and 60°C for 1 min. Transcript levels were calculated relative to the housekeeping gene GAPDH by using the ΔΔCT method. For cell-specific genotyping of MyD88, purified endothelial cells or microglia were lysed and genomic DNAs were isolated with ISOLATE II genomic DNA kits (Bioline). Primer sequences were as follows: MyD88: forward: 5′-GGCAACTAGAACAGACAGAC-3′, reverse: 5′-CAAATGCTGAAACTATAGG-3′; β-actin: forward: 5′-GGTTTAAGGGGAATCCCAGCA-3′, reverse: 5′- AGGGTGTAAAACG CAGCTCA-3′. PCR conditions were as follows: 3 min at 95°C, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 15 s, and extension at 72°C for 30 s. The reaction was finally incubated at 72°C for 1 min. PCR products were resolved on 1.5% agarose gels and imaged on a gel imager.
Protein Isolation and Enzyme-Linked Immunosorbent Assay
LPS- and PBS-treated animals were perfused with ice-cold PBS and their cortices were removed, snap-frozen in liquid nitrogen, and stored at −80°C. The samples were homogenized in ice-cold lysis buffer containing 20 mM HEPES (pH 7.9), 400 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 300 mM sucrose, and 5 µg/mL protease inhibitor cocktail (Sigma). The homogenates were centrifuged at 13,000g for 15 min at 4°C and the supernatants were stored at −80°C until use. Protein concentrations were determined using the Micro BCA Protein Assay (Thermo Scientific) per the manufacturer’s instructions. CXCL10 content in the samples was measured with an enzyme-linked immunosorbent assay (ELISA) kit (R&D systems).
Three-Dimensional EM
Three-dimensional (3D) EM was performed in a Carl Zeiss Sigma VP scanning electron microscope containing a 3View in-chamber ultramicrotome system and Gatan high sensitivity, low-kV backscattered electron detector (Gatan, England). Mice were perfused at one-day post-LPS or -PBS injections with cacodylate-buffered 2.5% glutaraldehyde and 4% paraformaldehyde. Brains were removed and 1 mm coronal slices were cut with a mouse brain matrix (Electron Microscopy Sciences). A 1 mm3 tissue block was further dissected from the motor cortex and treated with 2% OsO4-ferricyanide, followed sequentially by thiocarbohydrazide, aqueous OsO4 (2%), aqueous uranyl acetate, and Walton’s lead aspartate stain (CitationWalton, 1979) before they were embedded in Epon resin (all chemicals from Electron Microscopy Services). Serial images of the block face were generated by automated repeated cycles of cutting and scanning. Images were acquired with a 2.25 kV beam in high vacuum mode. Sets of 250 to 500 images at 75 nm steps (i.e., section thickness) were obtained at 8 to 10 nm per pixel resolution, producing images of 78 × 78 μm. Images were registered and corrected for histogram contrast, and derivative stacks were generated using ImageJ software with FIJI plug-in sets. 3D reconstructions were generated by using the 3D view plugin in FIJI. The volume was calculated as 78 μm × 78 μm × 75 μm × number of slices acquired. The number of villi was quantified and normalized to the volume of each reconstructed 3D image obtained from each mouse (number of villi/mm3 vessel cavity space). Three mice were used in the PBS group and four in the LPS group; values for each mouse are indicated by a circle in the figures.
Western Blots
Samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis performed with the XCell SureLock system (Invitrogen) using precast gradient gels; 30 μg of total protein was loaded in each lane. The gels were transferred to polyvinylidene difluoride membranes by using the DryBlot system (Invitrogen). The membranes were blocked for 1 hr at room temperature with 5% (w/v) nonfat-dried milk (Bio-Rad) in Tris-buffered saline with 0.1% Tween-20. The blots were then cut vertically into two halves in a way that the housekeeping protein GAPDH and the proteins of interest remained on each portion. The blots were placed in primary antibodies (GAPDH, 1:1000, Millipore; Claudin-5, 1:500, Thermo; Occludin, 1:500, Abcam) overnight at 4°C, washed and probed with horseradish peroxidase-tagged secondary antibodies (Jackson Immuno) for 1 hr at room temperature. Blots were mounted on the stage of a gel imager (Bio-Rad). Clarity ECL substrate (Bio-Rad) was spread over the entire blot and digital images of the chemiluminescent bands were captured with computer-optimized exposures. Quantification of the band intensity was performed with Image Lab software (Bio-Rad).
Mouse Model of Cryogenic Brain Injury and Behavioral Tests
Twenty-four hours after the last dose of LPS injection, cryogenic injury was initiated as a form of TBI with similarities to human TBI (CitationRaslan et al., 2012). This method was chosen due to the highly reproducible size and location of the lesion. Briefly, mice were anesthetized by IP injections of ketamine (100 mg/kg) and xylazine (10 mg/kg). The surgeon was blinded to the treatments (PBS and LPS) given to the mice. A 10 mm midline sagittal incision was made in the scalp. A unilateral aseptic cryogenic injury of the right parietal cortex was induced by applying a 3.5 mm diameter metallic probe, which was precooled in liquid nitrogen, on the skull for 15 s, resulting in blanching of the skull surface. The skin incision was closed and mice were allowed to recover. Neurological function was estimated by the beam-walk test, which measures the time it takes to cross a 30 cm long and 1 cm wide beam. Baseline performance was recorded 24 hr prior to injury and 24, 48, and 72 hr after the injury. The data are expressed as the fold change compared to the baseline. To quantify lesion volumes, 72 hr after the injury mice were deeply anesthetized and transcardially perfused with 4% paraformaldehyde, and brains were removed for assessment. Serial coronal brain sections (30 µm) were cut on a cryostat (Leica) and collected every 300 µm throughout the entire brain. This usually results in 10 to 15 sections depending on lesion volume variability between treatment groups. Sections were subsequently stained with Giemsa stain (Sigma) to visualize the lesions. Giemsa stains nuclei blue. Lesion areas were not stained because of the absence of viable cells with intact nuclei. Microscope slides were then scanned on a slide scanner (Microtek) and lesion volumes were calculated by multiplying the total lesion area by the section interval thickness and expressed in mm3.
Statistical Analysis
Prism for Windows (GraphPad Software) was used for unpaired Student’s t-tests or one-way analysis of variance (ANOVA) or two-way analysis of variance where appropriate. If the ANOVA produced a significant result, then Tukey’s post hoc method was applied for multiple comparisons among the individual groups. Behavioral data in the beam-walk test were analyzed by two-way ANOVA with least-significant difference (LSD) post hoc tests using SPSS software (IBM). Results are presented as mean ± SEM or otherwise stated. Statistical significance was defined as P < .05.
Results
IP Injections of LPS do not Alter BBB Permeability
LPS is highly heterogeneous, with varying lengths of polysaccharide chains and an estimated molecular weight that exceeds 4,000 g/mol (https://pubchem.ncbi.nlm.nih.gov/compound/LPS-core; accessed September 6, 2021). A single IP LPS injection results in elevated levels of LPS in blood circulation (CitationHuang et al., 2007), but does not disrupt the BBB (CitationBanks and Robinson, 2010). To determine whether daily IP LPS (1.0 mg/kg) injections over 4 days increase BBB permeability, BBB integrity was examined 24 hr after the fourth LPS injection using a sodium fluorescein (NaF) assay. NaF is a small (376 Da) fluorescence-emitting molecule that is one of the most widely used compounds for tracing BBB integrity (CitationSaunders et al., 2015). It only weakly binds to serum proteins, so a considerable portion of NaF remains free in the circulation, thereby enabling detection of subtle alterations in BBB permeability (CitationWolman et al., 1981). There was no evidence of BBB leakage 24 hr after four LPS injections, based upon similar levels of NaF fluorescence intensity recovered from brain tissues of control and LPS-injected mice (). Viral infection with the neurotropic coronavirus JMHV was used as a positive control for BBB disruption (; CitationPhares et al., 2014).
Figure 1 IP LPS injections activate CNS endothelial cells without compromising BBB integrity. (A) Compared to controls, four daily IP LPS injections (1.0 mg/kg) do not increase the fluorescent intensity of NaF transfused from the periphery to the brain. JHMV infection was used as a positive control. (B) Electron micrographs showing endothelial cell TJs in the cortex (TJs; arrowheads) were maintained in LPS-treated mice. N = 3 for PBS-treated mice and N = 4 for LPS-treated mice. (C) The endothelial TJ proteins claudin 5 and occludin were not significantly altered following IP LPS injections. (D) Flow cytometry analysis of CD31-positive brain endothelial cells following IP LPS injections. The gating strategy is shown with the top panels. The endothelial cell activation marker CD54 was significantly increased following four IP LPS injections, while the trend in increased CD105 and TLR4 expression did not reach statistical significance. (E) TLR4 expression in brain cells as examined by single-cell RNA sequencing queried from VascularSingleCells online database. *P < 0.05 by one-way ANOVA.
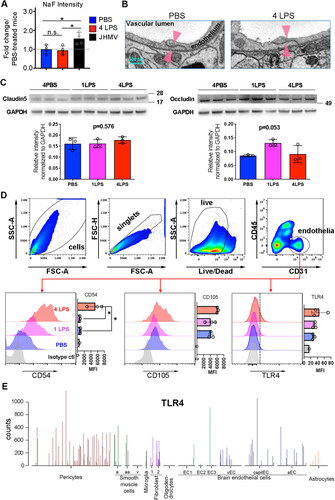
EM was used to assess the ultrastructural integrity of brain endothelial TJs in mice administered four LPS doses or three PBS injections. Examination of a total of 10 and 8 vessels, respectively, revealed no obvious damage to the TJs (). Fisher’s exact test was performed to statistically confirm that there was no difference in TJ damage between PBS- and LPS-treated mice (P > 0.9). Analysis of the major endothelial TJ proteins claudin-5 and occludin by Western blotting also did not reveal significant reductions in the levels of either protein in LPS-treated mice. Nevertheless, an apparent trend toward increased claudin-5 and occludin after one dose of LPS treatment may indicate enhanced protection of the BBB (). Together, these studies support that four daily IP LPS injections do not disrupt BBB integrity.
Brain Endothelial Cells are Activated by IP Injections of LPS
CD31-positive endothelial cells from control and LPS-treated mice were isolated 24 hr after the first or fourth LPS injection and examined for endothelial activation markers by flow cytometry (). The classical endothelial activation marker, CD54 (intercellular adhesion molecule-1), was already increased 24 hr after one and to a significantly higher level after four LPS injections relative to the PBS-treated group. CD105 (endoglin) and TLR4 (CitationLi et al., 1999) showed a trend toward increased expression 24 hr after the fourth LPS injection (), but mean fluorescence intensities (MFIs) did not reach statistical significance. TLR4 expression on brain endothelial cells was also queried using the VascularSingleCell online database (CitationVanlandewijck et al., 2018) with gene symbol TLR4. This tool provides transcriptomic information of TLR4 mRNA expression in the brain at a single-cell resolution. While not all brain endothelial cells were found to express TLR4, some are among the highest expressers in the brain (), corroborating that a subset of endothelial cells is capable of detecting LPS circulating in the bloodstream. These results are consistent with publications demonstrating that TLR4 is constitutively expressed at low levels on brain endothelial cells (CitationNagyoszi et al., 2010; CitationTang et al., 2018; CitationVanlandewijck et al., 2018), and can be further upregulated upon stimulation.
The flow cytometry results supporting endothelial cell activation were supplemented by enumeration of endothelial microvilli, a physical parameter of endothelial activation, by serial block face scanning EM (3D EM) after four doses of LPS treatment. The number of microvilli projecting from the luminal surface of brain vessels was significantly increased in LPS-treated mice ( to C). Taken together these results establish that brain vascular endothelial cells already exhibit activation after a single dose of IP-administered LPS, which is exacerbated after four injections.
Figure 2 IP LPS injections activate CNS endothelial cells. (A) Microvilli (arrowheads) were examined by EM. (B) 3D-reconstruction of microvilli in the vessel lumen as examined by 3D-EM. (C) Microvilli were significantly increased in LPS-treated mice. (D) Confocal microscopy shows that microglial activation (green) initially occurs in the vicinity of blood vessels (red). Scale bar = 25 μm. Arrowheads indicate the lengths of randomly selected longitudinal blood vessels analyzed for proximal CD31 and F4/80 staining. (E) Fluorescence intensity profiles demonstrate that activated microglia (green) line blood vessels (red). Arrowheads correspond to the arrowheads shown in panel (D). This result represents three independent experiments. Error bars represent SD. *P < 0.05 by Student’s t-test.
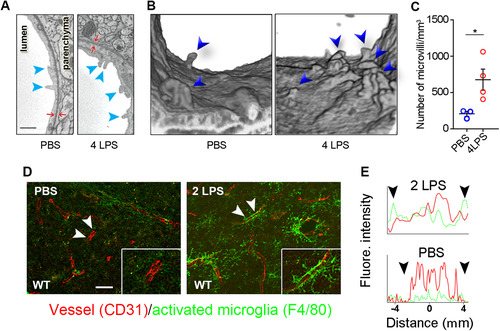
To assess whether activation of brain endothelial cells attracted activated microglia, brain sections from mice treated with two LPS doses to reflect intermediate levels of endothelial cell activation were analyzed for proximal localization of the vascular endothelial cell marker CD31 and the microglial activation marker F4/80 (). F4/80 staining was barely detectable in PBS-treated control mice (), but was readily detected adjacent to CD31-positive blood vessels in LPS-injected mice (, two LPS). The specialized microglial association with vessels is best demonstrated by viewing the vessels in a longitudinal orientation. In a normal brain, microglia with symmetrically oriented processes are restricted to small domains and the ends or small segments of individual processes may be in close opposition of blood vessels. In LPS-treated mice, microglial close to blood vessels extend long asymmetrically oriented processes that line blood vessels when they are viewed in longitudinal orientation (, arrowhead). For the assessment of coverage of endothelial cells by F480 staining microglia processes, the fluorescence intensity of both CD31 and F4/80 was measured along randomly selected blood vessels (indicated by a pair of arrowheads in ). F480 staining processes from activated microglia (green; ) closely oppose CD31-positive blood vessels (red; ) after two IP LPS injections. F4/80 staining was sparse in PBS-treated mice () and microglia did not longitudinally ensheath CD31-positive blood vessels. These differences are summarized by plotting the fluorescent intensity of both CD31 and F480 reactivity along a selected length of blood vessels (). Microglia located in close proximity to the vessels are among the first to be activated following IP LPS injections. These data support the recent report of LPS-induced microglial-vessel associations that protect BBB integrity (CitationHaruwaka et al., 2019).
Activated Endothelial Cells Upregulate Immune Modulators via MyD88 Signaling Pathways
To further explore transcriptional changes in brain-derived endothelial cells after a single- or 4-dose LPS regimen, brain endothelial cells were isolated 24 hr after one or four LPS or PBS injections, and RNA profiles were analyzed using NanoString analysis. The NanoString “Mouse inflammation CodeSet” comprises over 200 inflammation-related gene transcripts involved in signaling pathways, including that of TLR4 and MyD88. A comparison of gene transcripts using a distance matrix showed that the gene profile induced by a single LPS injection was distinct from that of PBS-injected controls (). Moreover, the heatmap demonstrates that a single LPS treatment significantly increased 17 transcripts, whereas 23 were significantly downregulated ( and extended data in Supplemental Table S1). Similarly, four LPS injections significantly upregulated 63 transcripts and downregulated 8 transcripts (extended data in Supplemental Table S1). A volcano scatter plot of differentially expressed gene transcripts in brain endothelial cells in one dose LPS- compared to PBS- treated mice revealed that Cxcl10, C3, Ccl2, Il1β, Cxcl2, and Cxcl1 were among the most significantly upregulated genes (P<.05; ). These levels were maintained or even further increased 24 hr after the fourth LPS injection ().
Figure 3 Cxcl10 is highly expressed by activated endothelial cells. (A) Distance matrix of endothelial cell expression data examined by NanoString analysis showing results from a single LPS injection. (B) Unsupervised hierarchical clustering of gene expressions by endothelial cells isolated from single-dose PBS- or LPS-treated mice. (C) Volcano dot plot of NanoString analysis showing that Cxcl10 is the most significantly upregulated transcript expressed by brain endothelial cells after one LPS injection. Transcripts above the dotted line have a P < .05. (D) Fold increases in individual transcripts following one or four LPS injections. *P < .05, ***P < .001 by Student’s t-test. (E) ELISA detected increased CXCL10 in brain extracts after one or four LPS injections. *P < .05, ***P < .001 by one-way ANOVA with Tukey’s post hoc test. N as indicated by individual circles. (F) STRING analysis supports MyD88 as an essential regulator of upregulated genes; Cxcl10 forms networks with other genes of interest. Error bars represent SD.
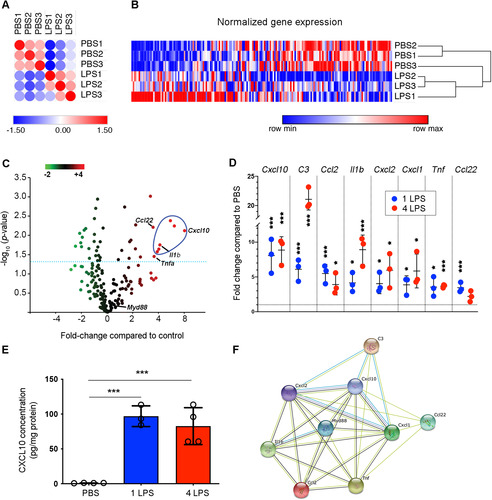
Cxcl10, the highest upregulated transcript, has been shown to activate microglia (CitationClarner et al., 2015; CitationTanuma et al., 2006) and facilitate their migration (CitationRappert et al., 2004). This established endothelial CXCL10 as a prime candidate for mediating LPS-induced microglial activation and neuroprotection. CXCL10 protein production was therefore initially confirmed by ELISA. CXCL10 was barely detectable in the control cortex (0.94 ± 0.17 pg/mg), but was significantly increased 24 hr after one (96.9 ± 14.9 pg/mg, P < .001) or four LPS injections (82.7 ± 26.5 pg/mg, P < .001; ). Furthermore, Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) analysis to visualize the functional association of highly upregulated endothelial transcripts following four LPS injections (CitationSzklarczyk et al., 2011) identified MyD88 as a regulator of the majority of these increased transcripts (). Importantly, CXCL10 was also revealed as a key factor associated with the function of the increased transcripts, supporting its functional relevance in this interactive network ().
Endothelial MyD88 Signaling is Essential for LPS-Induced Microglial Activation
To better understand the relative importance and interdependence of Myd88 signaling and CXCL10 production by endothelial cells in mediating microglia activation and neuroprotection endothelia-specific Myd88-deficient mice were generated by crossing Myd88fl/fl mice with Tie2eCre mice (CitationYu et al., 2014). The specificity and efficacy of the Cre recombinant reaction in endothelial cells, but not other cell types such as microglia, was assessed by PCR of the Myd88 gene in FACS-purified brain endothelial and microglia of naïve mice. The purity of the sorted cells was determined by flow cytometric analysis of a small portion of the sorted cells immediately after the sorting. A gate was set on the major population of these “enriched” cells to calculate the percentage of cells falling into this gate relative to the total cells acquired. Based on the cell population falling outside of the enriched gate, the purity of the sorted endothelial cells and microglia was ∼90% to ∼95% when compared to the cells prior to FACS sorting (). PCR analysis to assess the excision of the floxed Myd88 gene fragment in the genomic DNA revealed effective deletion from the endothelial cells isolated from Myd88fl/fl;Tie2eCre mice, but not Cre-negative littermate controls; furthermore, the Myd88 gene in microglia remained intact (). The residual Myd88 gene PCR product evident in the Cre+ endothelia () may be derived from the ∼10% nonendothelial population or incomplete excision.
Figure 4 Microglial activation is abolished when MYD88 is selectively deleted from endothelial cells. (A) Contour plots of the cells “before” and “after” FACS sorting. High-purity endothelial cells or microglia were obtained. (B) PCR reaction on FACS-sorted brain endothelial or microglial cells confirmed that the Myd88 gene is specifically and effectively removed from endothelial cells, but not from microglia. DNA gels of end-point PCR products are shown. (C) Brain endothelial Cxcl10 mRNA levels were not significantly increased in Tie2e-Cre+ mice after LPS treatment. Error bars represent SD. *P < .05; one-way ANOVA with Tukey’s post hoc test. N as indicated by individual circles. (D) Micrographs of IBA1 immunohistochemistry stain. Scale bar = 50 μm or 10 μm in the insets. Microglial activation depends upon the presence of endothelial MyD88. (E) Quantification of microglial activation. Error bars represent SD. **P < .01; one-way ANOVA with Tukey’s post hoc test. N as indicated by individual circles.
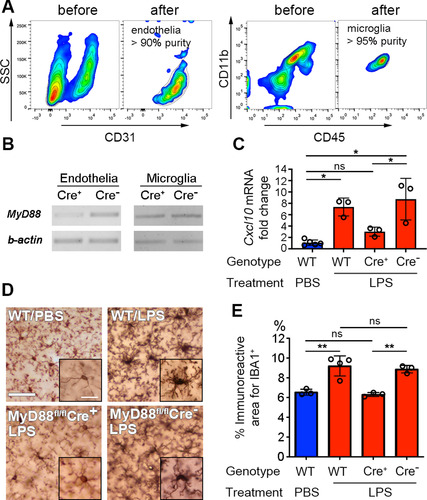
To directly prove the link between Myd88 signaling and CXCL10 production specifically in endothelial cells, endothelial cells were isolated by FACS from Myd88f/f;Tie2eCre+ mice and Cre-negative littermates 24 hr after four PBS or LPS injections. Quantitative RT-PCR showed that LPS failed to increase Cxcl10 mRNA transcription in Myd88-deficient endothelial cells (). The effect of impaired Myd88 signaling and Cxcl10 expression in endothelial cells on microglia activation was examined by the morphology of Iba-1-stained microglia using immunohistochemistry. Activated microglia display enlarged cell bodies and thickened primary processes, thereby increasing the area they occupy in their microenvironment (CitationChen et al., 2012). Interestingly, microglial activation was abolished in LPS-treated Myd88f/f;Tie2eCre+ mice (). These results establish that MyD88 signaling in brain endothelial cells is essential for increased endothelial CXCL10 expression and microglial activation in LPS-treated mice.
Endothelial CXCL10 is Essential for LPS-Induced Neuroprotection
Four daily IP LPS injections are prophylactic, protect against TBI induced by cryogenic injury, and depend upon microglia activation prior to induction of TBI (CitationChen et al., 2012, Citation2014). To investigate whether CXCL10 is necessary for the neuroprotection provided by LPS preconditioning, WT and Cxcl10−/− mice were subjected to cryogenic injury and neurobehavioral tests ( to C). WT mice pretreated with four LPS injections had significantly smaller lesion volumes than those treated with PBS (; 7.6 ± 2.3 mm3 vs. 10.6 ± 2.2 mm3, P < .05). However, this protection was abolished in Cxcl10−/− mice (; 9.2 ± 1.1 mm3 vs. 8.6 ± 3.1 mm3). The beam-walk test was used to determine whether LPS neuroprotection and loss of CXCL10 altered motor performance (). Following cryogenic brain injury, the beam crossing time was prolonged to approximately 8.5-fold over baseline for both PBS-treated WT and Cxcl10−/− mice. However, while LPS preconditioning significantly decreased beam crossing time to 2.5-fold over baseline in WT mice (P < .01; ), it had no effect on beam crossing time in Cxcl10−/− mice (). These data establish that CXCL10 is required for LPS-induced neuroprotection and improved motor performance.
Figure 5 Neuroprotection is diminished in Cxcl10−/− mice. (A) Representative images of cryogenic lesions in WT and Cxcl10−/− mice. Lesions appear as discolored regions and are demarcated by dotted lines. (B) Lesions volumes were reduced in LPS-treated WT mice, but not in LPS-treated Cxcl10−/− mice. Error bars represent SD. *P < .05, one-way ANOVA with Tukey’s post hoc test. (C) Beam-crossing behavior test of WT and Cxcl10−/− mice following brain injury. **P < .01 by two-way ANOVA test. (D) Representative images of microglial activation in PBS- and LPS-treated Cxcl10−/− mice. (E) The percentage of cortical area occupied by microglia was increased in LPS-treated WT mice, but not in LPS-treated Cxcl10−/− mice. **P < .01, one-way ANOVA with Tukey’s post hoc test. N is indicated by individual circles. (F) Confocal micrographs of neurons (red), microglia (blue), and GAD-positive inhibitory synapses (green) in PBS- and LPS-treated Cxcl10−/− mice. The neuronal circumference occupied by GAD-positive synapses was reduced in WT mice, but not in Cxcl10−/− mice. Error bars represent SD. **P < 0.01, one-way ANOVA with Tukey’s post hoc test. N is indicated by individual circles.
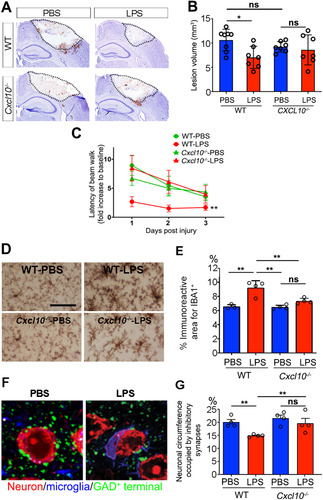
Microglial activation was monitored by examining Iba-1 immunoreactivity in Cxcl10−/− mice (). The area occupied by microglia was significantly increased in LPS-injected WT mice compared to PBS controls (; 9.17 ± 1.04% vs. 6.53 ± 0.31%, P < .01). By contrast, LPS-induced microglial activation was abolished in Cxcl10−/− mice (; 7.32 ± 0.38% vs. 6.53 ± 0.31%). A prior study demonstrated that LPS-induced neuroprotection was associated with activated microglia ensheathing neuronal somas and displacing GAD-positive presynaptic terminals 24 hr after the fourth LPS injection (CitationChen et al., 2014). To assess the disruption of this protective mechanism in the absence of CXCL10, the neuronal surface area occupied by GAD-positive puncta was compared in WT and Cxcl10−/− mice (). In contrast to WT mice, no reductions in GABAergic synapses were detected in Cxcl10−/− mice (), supporting that CXCL10 is required for microglial activation and displacement of GAD inhibitory terminals.
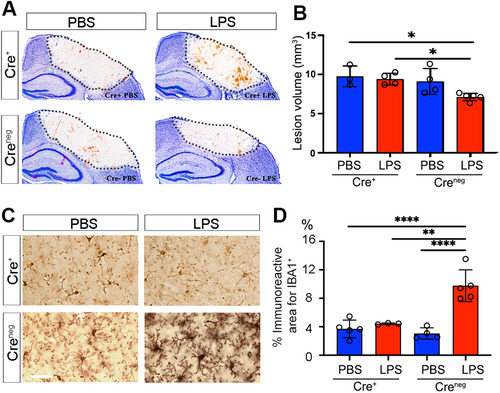
Finally, to specifically confirm the contribution of endothelial-derived CXCL10 to LPS-induced neuroprotection, Cxcl10fl/fl mice were crossed with Tie2eCre mice. Both Cre-positive mice and their Cre-negative littermates were given PBS or LPS for four consecutive days and subsequently subjected to cryogenic brain injury. A comparison of cryogenic lesion sizes by Giemsa staining () showed that lesion volume was significantly smaller in LPS-treated mice with intact CXCL10 compared to abrogated endothelial CXCL10 (; 7.12 ± 0.46 mm3 in Cre− vs. 9.4 ± 0.75 mm3 in Cre+ mice, P < .001). LPS failed to induce microglial activation in Cxcl10fl/fl;Tie2eCre mice. IBA1 reactive areas were similar in Cxcl10fl/fl;Tie2eCre, PBS Cre+, and PBS Cre− control mice, which were all significantly lower than in LPS-treated Cre− mice (). The lack of microglia activation was consistent with results in Myd88fl/fl;Tie2eCre mice () and global Cxcl10−/− mice (). These data confirm the indispensable role of endothelia-specific CXCL10 in LPS-mediated neuroprotection and microglial activation.
Figure 6 Neuroprotection and microglial activation are abolished when CXCL10 is selectively abolished from brain endothelial cells. (A) Representative images of cryogenic lesions in PBS- and LPS-treated Cre+ and Creneg Cxcl10fl/fl;Tie2eCre mice visualized by Giemsa stain. Lesions appear as discolored regions and are demarcated by dotted lines. (B) Lesion volumes were reduced in LPS-treated Creneg mice, but not in LPS-treated Cre+ mice. Error bars represent SD. *P < .05 by one-way ANOVA with Tukey’s post hoc test. (C) Cortical microglia in PBS-treated and LPS-treated Creneg and Cre+ mice. (D) The percentage of cortical area occupied by microglia was increased in LPS-treated Creneg, but not in LPS-treated Cre+ mice. Error bars represent SD. **P < .01, ****P < .0001, one-way ANOVA with Tukey’s post hoc test. N as indicated by individual circles.
Discussion
Transient neuroprotection can be induced by a variety of subthreshold noxious stimuli. Such treatments are collectively referred to as preconditioning, a phenomenon in which the brain transiently protects itself against future injury by adapting to low doses of noxious insults (CitationCadet and Krasnova, 2009; CitationShpargel et al., 2008). LPS preconditioning transiently protects the brain against focal cerebral ischemia (CitationDawson et al., 1999), seizures (CitationMirrione et al., 2010), and TBI (CitationChen et al., 2014). The results herein provide compelling evidence that the initial site-of-action of LPS-mediated neuroprotection involves TLR4 receptors expressed on the luminal surface of brain vascular endothelial cells.
The data support that MyD88 signaling in endothelial cells is essential for LPS-induced neuroprotection and that endothelial expression of the chemokine CXCL10 is a key player in this process. Specifically, the results demonstrate that (1) low-dose LPS injected into the peritoneum activates brain endothelial cells without altering BBB integrity; (2) activated endothelial cells express high levels of gene transcripts that encode cytokines and chemokines; and (3) MyD88-dependent endothelial signaling and endothelial CXCL10 are essential for LPS-induced neuroprotection, microglial activation, and microglial-mediated stripping of inhibitory synapses. This study has thus identified a novel pathway by which the vasculature provides protection to the CNS via release of endothelial CXCL10 and activation of proximal microglia.
MyD88 signaling was identified as the key endothelial cell pathway induced by IP injection of LPS (). Effective deletion of the Myd88 gene from brain endothelial cells () failed to activate microglia or to significantly increase CXCL10 following four LPS injections (). The lack of microglial activation in Myd88fl/fl;Tie2e-Cre mice () provides additional evidence that LPS-administered IP does not directly activate Myd88-sufficient microglia. LPS-mediated TLR4 signaling can also activate TIR-domain-containing adaptor-inducing IFNβ (TRIF), which is a MyD88-independent pathway that exhibits a delayed response (CitationAkira et al., 2006) to LPS stimulation. The TRIF-mediated pathway translocates the nuclear factor IRF3, initiating the production of IFN and IFN-induced genes, including CXCL10. While it cannot be excluded that TRIF signaling plays a role in the LPS paradigm, it does not activate microglia in Myd88fl/fl;Tie2e-Cre mice. It can also not be fully ruled out that peripheral blood vessels participate in LPS-induced neuroprotection as Tie2e is also expressed by non-CNS endothelial cells. Future studies using Slco1c1-Cre mice could directly address this possibility. Slco1c1 is expressed by brain endothelial cells and astrocytes, but not by non-CNS endothelial cells (CitationRidder et al., 2011).
CXCL10 was the highest upregulated gene transcript in LPS-activated endothelial cells. Specific elimination of CXCL10 from endothelial cells resulted in both the loss of LPS-mediated neuroprotection from traumatic cryo-injury and microglia activation (). CXCL10 can reduce GABA receptor expression and GABAergic synaptic activity in vitro (CitationBajova et al., 2008; CitationCho et al., 2009). In addition, a subpopulation of cortical microglia that expresses GABAB receptors selectively prunes cortical inhibitory synapses during development (CitationFavuzzi et al., 2021). These data raise the possibility that CXCL10 has a direct effect on both neuronal and microglial GABA receptor subunit expression. CXCR3, the cognate receptor for CXCL10, is expressed by neurons (CitationPetrisko et al., 2020) and microglia (CitationRappert et al., 2004). It remains to be determined if endothelial CXCL10 directly modulates neuronal and microglial GABA receptor subunits, and whether activated microglia are causing or responding to GABA receptor modulations.
Microglial activation correlates with LPS-induced neuroprotection, and both were abolished in mice deficient in endothelial CXCL10. Microglia continuously monitor neuronal health. Following CNS injury, microglial processes associate with neuronal somata and provide neuroprotection (CitationCserep et al., 2020). This neuroprotection requires microglial expression of the purinoreceptor, P2Y12, neuronal release of ATP as a “find me signal,” neuronal membrane clustering of the voltage-gated potassium channel Kv2.1 (CitationCserep et al., 2020) as exocytotic and endocytotic hubs (CitationDeutsch et al., 2012; CitationFeinshreiber et al., 2009; CitationFox et al., 2015), and neuronal mitochondrial fragmentation and dysfunction, which is responsible for increased extracellular adenosine triphosphate (ATP; CitationCserep et al., 2020). The data herein support an additional microglial–neuronal association where microglia physically associate with neuronal cell bodies in response to the extrinsic threat of a bacterial infection. This “damage predictor” function of microglial–neuronal interactions thus supplements their direct “damage sensing” function. Mitigation of “damage prediction” increases neuronal cell loss in rodent models of focal cerebral ischemia (CitationDawson et al., 1999), seizures (CitationMirrione et al., 2010), and TBI (CitationChen et al., 2014). While the molecular mechanisms responsible for the physical association between neurons and “damage predicting” microglia remain to be determined, this association increases neuronal expression of survival and neurotrophic proteins through CREB-mediated transcription and decreases apoptotic signals through phosphorylation of BAD (CitationChen et al., 2014). While endothelial CXC10 expression is essential for microglial damage prediction and neuroprotection, the underlying mechanisms may require participation of additional CNS cell types and molecules.
Another interesting observation is the increase in complement proteins following four daily LPS injections (Extended data). It has been shown that complement, C1q, actively participates in microglia-mediated synaptic pruning (CitationPaolicelli et al., 2011; CitationSchafer et al., 2012). Complement may be involved in the transient displacement of inhibitory synapses following LPS treatment. This displacement appears to be reversible and does not result in microglial-mediated phagocytosis of the presynaptic terminal (CitationChen et al., 2014) raising the possibility of novel complement functions in LPS-induced microglial activation.
How LPS-activated endothelial cells communicate with microglia and/or neurons to mediate neuroprotection remains to be further explored. Short-distance microglial mobility is likely to occur as microglia ensheath neurons and/or oppose blood vessels. However, microglial proliferation after LPS treatment or following TBI has not been observed in this model, which was discussed in depth by CitationChen et al. (2014). Microglia constantly monitor their surroundings, including the health of neurons through multiple receptor–ligand interactions, including CX3CR1–CX3CL1. CX3CL1 can also be expressed by activated endothelial cells, potentially providing a communication pathway to microglia in addition to P2RY12 signaling. Interestingly, microglia have been shown to contribute both to the repair of injured BBB or direct BBB injury (CitationHaruwaka et al., 2019; CitationLou et al., 2016). Similarly, TREM2 signaling exerts neuroprotective effects in CNS neurodegenerative diseases and injury (recently reviewed in CitationPuntambekar et al., 2018). Loss-of-function mutations in Trem2 are associated with the diminished ability of phagocytic function and thus the ability to clear myelin debris, β-amyloid, and cellular debris, as well as increased induction of proinflammatory mediators, which can potentially affect TBI pathology at both early and chronic phases. The involvement of these receptors in the LPS protective paradigm deserves future attention.
In summary, activation of brain endothelial TLR4 transiently protects the cerebral cortex from traumatic injury. It is likely that this neuroprotective pathway evolved as a response to circulating gram-negative bacteria, as their entrance into the CNS would have traumatic effects. Activation of endothelial TLR4 therefore provides a detection signal that preconditions the brain for a possible parenchymal infection. While LPS is not an attractive therapeutic agent for neuroprotection in humans, the use of this model provides proof-of-principle that activation of TLR4 receptors located on the luminal surface of brain endothelial cells can protect the brain from acute injury by engaging endothelial MyD88 signaling and endothelial CXCL10 as essential components. Targeting activation of brain endothelial receptors provides an attractive and practical approach for inducing transient neuroprotection and for investigating the role of neurovascular units and neuronal–glial interactions in neuroprotection.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ORCID iDs
Mynor J. Mendez https://orcid.org/0000-0001-7899-0751
Bruce D. Trapp https://orcid.org/0000-0001-6682-3737
sj-docx-1-asn-10.1177_17590914221146365 - Supplemental material for Neuroprotection by Preconditioning in Mice is Dependent on MyD88-Mediated CXCL10 Expression in Endothelial Cells
Download MS Word (27.5 KB)sj-docx-2-asn-10.1177_17590914221146365 - Supplemental material for Neuroprotection by Preconditioning in Mice is Dependent on MyD88-Mediated CXCL10 Expression in Endothelial Cells
Download MS Word (19.9 KB)Acknowledgments
We thank Dr. Christopher L. Nelson for the scientific discussion and editorial support. We thank Ms. Jennifer Powers for FACS sorting and Ms. Amanda Wilson for animal husbandry.
Supplemental Material
Supplemental Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ZC was supported by a fellowship from the American Epilepsy Society. BTB was partly supported by an MSTP training grant (T32GM007250). CB is supported by grants from the National Institutes of Health (R01NS086299 and R01NS110700). This work was supported by a grant from the National Institutes of Health (R35NS090739 to BDT).
Supplemental material
Supplemental material for this article is available online.
Related Research Data
References
- Akira S., Uematsu S., Takeuchi O. (2006). Pathogen recognition and innate immunity. Cell, 124(4), 783–801. https://doi.org/10.1016/j.cell.2006.02.015
- Bajova H., Nelson T. E., Gruol D. L. (2008). Chronic CXCL10 alters the level of activated ERK1/2 and transcriptional factors CREB and NF-kappaB in hippocampal neuronal cell culture. Journal of Neuroimmunology, 195(1–2), 36–46. https://doi.org/10.1016/j.jneuroim.2008.01.003
- Banks W. A., Robinson S. M. (2010). Minimal penetration of lipopolysaccharide across the murine blood–brain barrier. Brain Behavior and Immunity, 24(1), 102–109. https://doi.org/10.1016/j.bbi.2009.09.001
- Cadet J. L., Krasnova I. N. (2009). Cellular and molecular neurobiology of brain preconditioning. Molecular Neurobiology, 39(1), 50–61. https://doi.org/10.1007/s12035-009-8051-6
- Chen Z., Jalabi W., Hu W., Park H. J., Gale J. T., Kidd G. J., Bernatowicz R., Gossman Z. C., Chen J. T., Dutta R., Trapp B. D. (2014). Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nature Communications, 5, 4486. https://doi.org/10.1038/ncomms5486
- Chen Z., Jalabi W., Shpargel K. B., Farabaugh K. T., Dutta R., Yin X., Kidd G. J., Bergmann C. C., Stohlman S. A., Trapp B. D. (2012). Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. Journal of Neuroscience, 32(34), 11706–11715. https://doi.org/10.1523/JNEUROSCI.0730-12.2012
- Chen Z., Trapp B. D. (2016). Microglia and neuroprotection. Journal of Neurochemistry, 136(S1), 10–17. https://doi.org/10.1111/jnc.13062
- Cho C. F., Wolfe J. M., Fadzen C. M., Calligaris D., Hornburg K., Chiocca E. A., Agar N. Y. R., Pentelute B. L., Lawler S. E. (2017). Blood–brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nature Communications, 8, 15623. https://doi.org/10.1038/ncomms15623
- Cho J., Nelson T. E., Bajova H., Gruol D. L. (2009). Chronic CXCL10 alters neuronal properties in rat hippocampal culture. Journal of Neuroimmunology, 207(1–2), 92–100. https://doi.org/10.1016/j.jneuroim.2008.12.007
- Clarner T., Janssen K., Nellessen L., Stangel M., Skripuletz T., Krauspe B., Hess F. M., Denecke B., Beutner C., Linnartz-Gerlach B., Neumann H., Vallieres L., Amor S., Ohl K., Tenbrock K., Beyer C., Kipp M. (2015). CXCL10 Triggers early microglial activation in the cuprizone model. Journal of Immunology, 194(7), 3400–3413. https://doi.org/10.4049/jimmunol.1401459
- Cserep C., Pósfai B., Lénárt N., Fekete R., László Z.I., Lele Z., Orsolits B., Molnár G., Heindl S., Schwarcz A.D., Ujvári K., Környei Z., Tóth K., Szabadits E., Sperlágh B., Baranyi M., Csiba L., Hortobágyi T., Maglóczky Z., …, Dénes Á. (2020). Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science (New York, N.Y.), 367(6477), 528–537. https://doi.org/10.1126/science.aax6752
- Dawson D. A., Furuya K., Gotoh J., Nakao Y., Hallenbeck J. M. (1999). Cerebrovascular hemodynamics and ischemic tolerance: Lipopolysaccharide-induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. Journal of Cerebral Blood Flow and Metabolism, 19(6), 616–623. https://doi.org/10.1097/00004647-199906000-00004
- Deutsch E., Weigel A. V., Akin E. J., Fox P., Hansen G., Haberkorn C. J., Loftus R., Krapf D., Tamkun M. M. (2012). Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Molecular Biology of the Cell, 23(15), 2917–2929. https://doi.org/10.1091/mbc.e12-01-0047
- Faden A. I., Stoica B. (2007). Neuroprotection: Challenges and opportunities. Archives of Neurology, 64(6), 794–800. https://doi.org/10.1001/archneur.64.6.794
- Favuzzi E., Huang S., Saldi G. A., Binan L., Ibrahim L. A., Fernandez-Otero M., Cao Y., Zeine A., Sefah A., Zheng K., Xu Q., Khlestova E., Farhi S. L., Bonneau R., Datta S. R., Stevens B., Fishell G. (2021). GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell, 184(15), 4048–4063. e4032. https://doi.org/10.1016/j.cell.2021.06.018
- Feinshreiber L., Singer-Lahat D., Ashery U., Lotan I. (2009). Voltage-gated potassium channel as a facilitator of exocytosis. Annals of the New York Academy of Sciences, 1152, 87–92. https://doi.org/10.1111/j.1749-6632.2008.03997.x
- Fleming J. O.,, Trousdale M. D., el-Zaatari F. A., Stohlman S. A., Weiner L. P. (1986). Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. Journal of Virology, 58(3), 869–875. https://doi.org/10.1128/jvi.58.3.869-875.1986
- Fox P. D., Haberkorn C. J., Akin E. J., Seel P. J., Krapf D., Tamkun M. M. (2015). Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. Journal of Cell Science, 128(11), 2096–2105. https://doi.org/10.1242/jcs.166009
- Hardingham G. E., Arnold F. J., Bading H. (2001). A calcium microdomain near NMDA receptors: On switch for ERK-dependent synapse-to-nucleus communication. Nature Neuroscience, 4(6), 565–566. https://doi.org/10.1038/88380
- Haruwaka K., Ikegami A., Tachibana Y., Ohno N., Konishi H., Hashimoto A., Matsumoto M., Kato D., Ono R., Kiyama H., Moorhouse A. J., Nabekura J., Wake H. (2019). Dual microglia effects on blood–brain barrier permeability induced by systemic inflammation. Nature Communications, 10(1), 5816. https://doi.org/10.1038/s41467-019-13812-z
- Huang H., Liu T., Rose J. L., Stevens R. L., Hoyt D. G. (2007). Sensitivity of mice to lipopolysaccharide is increased by a high saturated fat and cholesterol diet. Journal of Inflammation (London), 4, 22. https://doi.org/10.1186/1476-9255-4-22
- Li D. Y., Sorensen L. K., Brooke B. S., Urness L. D., Davis E. C., Taylor D. G., Boak B. B., Wendel D. P. (1999). Defective angiogenesis in mice lacking endoglin. Science (New York, N.Y.), 284(5419), 1534–1537. https://doi.org/10.1126/science.284.5419.1534
- Lou N., Takano T., Pei Y., Xavier A. L., Goldman S. A., Nedergaard M. (2016). Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proceedings of the National Academy of Sciences of the United States of America, 113(4), 1074–1079. https://doi.org/10.1073/pnas.1520398113
- Mirrione M. M., Konomos D. K., Gravanis I., Dewey S. L., Aguzzi A., Heppner F. L., Tsirka S. E. (2010). Microglial ablation and lipopolysaccharide preconditioning affects pilocarpine-induced seizures in mice. Neurobiology of Disease, 39(1), 85–97. https://doi.org/10.1016/j.nbd.2010.04.001
- Nagyoszi P., Wilhelm I., Farkas A. E., Fazakas C., Dung N. T., Hasko J., Krizbai I. A. (2010). Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochemistry International, 57(5), 556–564. https://doi.org/10.1016/j.neuint.2010.07.002
- Paolicelli R. C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T. A., Guiducci E., Dumas L., Ragozzino D., Gross C. T. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science (New York, N.Y.), 333(6048), 1456–1458. https://doi.org/10.1126/science.1202529
- Petrisko T. J., Bloemer J., Pinky P. D., Srinivas S., Heslin R. T., Du Y., Setti S. E., Hong H., Suppiramaniam V., Konat G. W., Reed M. N. (2020). Neuronal CXCL10/CXCR3 axis mediates the induction of cerebral hyperexcitability by peripheral viral challenge. Frontiers in Neuroscience, 14, 220. https://doi.org/10.3389/fnins.2020.00220
- Phares T. W., DiSano K. D., Stohlman S. A., Bergmann C. C. (2014). Progression from IgD + IgM + to isotype-switched B cells is site specific during coronavirus-induced encephalomyelitis. Journal of Virology, 88(16), 8853–8867. https://doi.org/10.1128/JVI.00861-14
- Puntambekar S. S., Saber M., Lamb B. T., Kokiko-Cochran O. N. (2018). Cellular players that shape evolving pathology and neurodegeneration following traumatic brain injury. Brain Behavior and Immunity, 71, 9–17. https://doi.org/10.1016/j.bbi.2018.03.033
- Rappert A., Bechmann I., Pivneva T., Mahlo J., Biber K., Nolte C., Kovac A. D., Gerard C., Boddeke H. W., Nitsch R., Kettenmann H. (2004). CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. Journal of Neuroscience, 24(39), 8500–8509. https://doi.org/10.1523/JNEUROSCI.2451-04.2004
- Raslan F., Albert-Weissenberger C., Ernestus R. I., Kleinschnitz C., Siren A. L. (2012). Focal brain trauma in the cryogenic lesion model in mice. Experimental & Translational Stroke Medicine, 4, 6. https://doi.org/10.1186/2040-7378-4-6
- Ridder D. A., Lang M. F., Salinin S., Roderer J. P., Struss M., Maser-Gluth C., Schwaninger M. (2011). TAK1 In brain endothelial cells mediates fever and lethargy. Journal of Experimental Medicine, 208(13), 2615–2623. https://doi.org/10.1084/jem.20110398
- Sangaran P. G., Ibrahim Z. A., Chik Z., Mohamed Z., Ahmadiani A. (2021). LPS preconditioning attenuates apoptosis mechanism by inhibiting NF-kappaB and caspase-3 activity: TLR4 pre-activation in the signaling pathway of LPS-induced neuroprotection. Molecular Neurobiology, 58(5), 2407–2422. https://doi.org/10.1007/s12035-020-02227-3
- Saunders N. R., Dziegielewska K. M., Mollgard K., Habgood M. D. (2015). Markers for blood-brain barrier integrity: How appropriate is Evans blue in the twenty-first century and what are the alternatives? Frontiers in Neuroscience, 9, 385. https://doi.org/10.3389/fnins.2015.00385
- Schafer D. P., Lehrman E. K., Kautzman A. G., Koyama R., Mardinly A. R., Yamasaki R., Ransohoff R. M., Greenberg M. E., Barres B. A., Stevens B. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74(4), 691–705. https://doi.org/10.1016/j.neuron.2012.03.026
- Shpargel K. B., Jalabi W., Jin Y., Dadabayev A., Penn M. S., Trapp B. D. (2008). Preconditioning paradigms and pathways in the brain. Cleveland Clinical Journal of Medicine, 75(Suppl 2), S77–S82. https://doi.org/10.3949/ccjm.75.suppl_2.s77
- Stevens S. L., Leung P. Y., Vartanian K. B., Gopalan B., Yang T., Simon R. P., Stenzel-Poore M. P. (2011). Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. Journal of Neuroscience, 31, 8456–8463.
- Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., von Mering C. (2011). The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Research, 39(Database issue), D561–D568. https://doi.org/10.1093/nar/gkq973
- Tang Z., Guo D., Xiong L., Wu B., Xu X., Fu J., Kong L., Liu Z., Xie C. (2018). TLR4/PKCalpha/occludin Signaling pathway may be related to blood–brain barrier damage. Molecular Medicine Reports, 18(1), 1051–1057. https://doi.org/10.3892/mmr.2018.9025
- Tanuma N., Sakuma H., Sasaki A., Matsumoto Y. (2006). Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathologica, 112(2), 195–204. https://doi.org/10.1007/s00401-006-0083-7
- Vanlandewijck M., He L., Mae M. A., Andrae J., Ando K., Del Gaudio F., Nahar K., Lebouvier T., Lavina B., Gouveia L., Sun Y., Raschperger E., Rasanen M., Zarb Y., Mochizuki N., Keller A., Lendahl U., Betsholtz C. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature, 554(7693), 475–480. https://doi.org/10.1038/nature25739
- Vaure C., Liu Y. (2014). A comparative review of toll-like receptor 4 expression and functionality in different animal species. Frontiers in Immunology, 5, 316. https://doi.org/10.3389/fimmu.2014.00316
- Walton J. (1979). Lead asparate, an en bloc contrast stain particularly useful for ultrastructural enzymology. Journal of Histochemistry and Cytochemistry, 27(10), 1337–1342. https://doi.org/10.1177/27.10.512319
- Wolman M., Klatzo I., Chui E., Wilmes F., Nishimoto K., Fujiwara K., Spatz M. (1981). Evaluation of the dye-protein tracers in pathophysiology of the blood–brain barrier. Acta Neuropathologica, 54(1), 55–61. https://doi.org/10.1007/BF00691332
- Yu M., Zhou H., Zhao J., Xiao N., Roychowdhury S., Schmitt D., Hu B., Ransohoff R. M., Harding C. V., Hise A. G., Hazen S. L., DeFranco A. L., Fox P. L., Morton R. E., Dicorleto P. E., Febbraio M., Nagy L. E., Smith J. D., Wang J. A., Li X. (2014). MyD88-dependent interplay between myeloid and endothelial cells in the initiation and progression of obesity-associated inflammatory diseases. Journal of Experimental Medicine, 211(5), 887–907. https://doi.org/10.1084/jem.20131314