
Abstract
Aberrant phosphorylation and subsequent aggregation of the trans-activation response (TAR) element DNA binding protein 43 (TDP-43) is a common feature of multiple neurodegenerative disorders and contributes to disease severity. Here, we investigated whether pathologic phosphorylation of TDP-43 (pTDP-43) is a hallmark of human immunodeficiency virus (HIV)- infected brains. We evaluated pTDP-43 immunoreactivity and TDP-43 kinases in HIV-infected (HIV + ) and seronegative post-mortem brain samples. We then used an inducible transgenic mouse model of the HIV-1 protein Tat and primary neuronal cultures, to decipher the underlying mechanism of the proteinopathy. Since opioid use disorder (OUD) can exaggerate HIV neuropathology, we explored interactions between HIV-1 Tat and morphine, a prototypical opioid, for all outcome measures. Cytoplasmic pTDP-43 and TDP-43 immunoreactivities were increased in neurons of the basal ganglia of post-mortem, HIV+ human tissues compared to seronegative controls. An evaluation of TDP-43 kinases revealed an increase in the levels of cytoplasmic casein kinase 2 (CK2) in HIV-positive human tissues but not CK1δ. There was a significant positive correlation between pTDP-43 and CK2 levels. Eight weeks of Tat induction and 2-week subcutaneous morphine exposure (10–40 mg/kg, increasing by 10 mg/kg/b.i.d.) independently produced similar outcomes for cytoplasmic pTDP-43 and CK2 levels in the mouse striatum. In primary, mouse striatal neuronal cultures, co-exposure to Tat and morphine for 24 h increased pTDP-43 levels and CK2 activity. Co-treatment with the CK2 antagonist CX-4945 prevented the Tat- and morphine-induced increases in pTDP-43 levels. Our results demonstrate that CK2 may be a viable therapeutic target for treating pTDP-43 proteinopathy in neuroHIV and OUD.
Summary Statement
HIV/HIV-1 Tat and morphine independently increase pathologic phosphorylation of TAR DNA binding protein 43 in the striatum. HIV- and opioid-induced pathologic phosphorylation of TAR DNA binding protein 43 may involve enhanced CK2 activity and protein levels.
Introduction
Current advancements in antiretroviral therapies for HIV (cART) have greatly improved the life expectancy of HIV-infected individuals, however, the persistence of mild to moderate forms of neurocognitive disorders (Human immunodeficiency virus (neuroHIV)) negatively impacts the quality of life of infected individuals. The aggregation of pathologic proteins in neurons has become a hallmark of many neurodegenerative disorders (CitationJucker & Walker, 2018; CitationPeng et al., 2020), including neuroHIV (CitationBhargavan et al., 2021; CitationBrown et al., 2014), and has prompted the development of new therapeutic strategies to limit or prevent specific proteinopathies. The present study investigated whether aberrant phosphorylation and neuronal aggregation of TAR DNA binding protein 43 (TDP-43), an emerging characteristic of a number of neurodegenerative disorders (CitationJo et al., 2020), occurs in HIV-exposed brains.
TDP-43 is a DNA/RNA binding protein that is ubiquitously expressed by cells and plays a crucial role in regulating gene expression (reviewed by (CitationJo et al., 2020)). TDP-43 was initially identified as a host cell factor that regulates HIV-1 gene expression in immune cells (CitationOu et al., 1995). TDP-43 has a normal tendency to form reversible dimers and oligomers, and this is enhanced by certain mutations within the N-terminal domain of TDP-43 (CitationJiang et al., 2016, Citation2017; CitationTsoi et al., 2017). While TDP-43 dimerization/oligomerization has been hypothesized to encourage aggregate formation (CitationTsoi et al., 2017), others have argued that TDP-43 self-dimerization protects against the deposition of TDP-43 cytoplasmic aggregates (CitationJiang et al., 2017). Aberrant TDP-43 aggregates have been identified in neurodegenerative disorders including amyotrophic lateral sclerosis, frontotemporal dementia, Parkinson’s disease, and Alzheimer’s disease (CitationGoossens et al., 2015; CitationHuang et al., 2020; CitationJo et al., 2020; CitationJosephs et al., 2014; CitationNelson et al., 2019; CitationRayaprolu et al., 2013; CitationSteinacker et al., 2019; CitationTomé et al., 2020). TDP-43 is normally localized to the nucleus, but pathologic conditions often involve nuclear-to-cytoplasmic translocation commonly termed mislocalization (CitationAlquezar et al., 2016; CitationBarmada et al., 2010; CitationGuerrero et al., 2019a; CitationNonaka et al., 2016), aberrant phosphorylation of key residues (CitationAlquezar et al., 2016; CitationGuerrero et al., 2019b; CitationNonaka et al., 2016; CitationWu et al., 2020), genetic mutations, and polyubiquitination (CitationNeumann et al., 2007). In the cytoplasm, hyperphosphorylation of TDP-43 at the Ser409/410 moieties by casein kinase 1δ (CK1δ) (CitationAlquezar et al., 2016; CitationHasegawa et al., 2008; CitationNonaka et al., 2016) or casein kinase 2 (CK2) (CitationCarlomagno et al., 2014; CitationHasegawa et al., 2008), contributes to pathologic aggregate formation (CitationBrady et al., 2011). Aggregation of TDP-43 in neurons can lead to mitochondrial dysfunction (CitationWang et al., 2016, Citation2017) and cell death (CitationAlquezar et al., 2016; CitationNonaka et al., 2016). In Alzheimer’s disease, abnormal phosphorylation and cytoplasmic inclusions are observed in patients who present with mild cognitive impairments (CitationTremblay et al., 2011), which is also a characteristic feature of neuroHIV in the cART era. TDP-43 aggregation in Alzheimer’s disease also contributes to the severity of neurocognitive impairments presented by the affected individuals [e.g., (CitationJosephs et al., 2014)].
A recent study reported an enhanced TDP-43 expression in cortical neurons of post-mortem HIV-infected brains (CitationDouville & Nath, 2017). Increased expression of the human endogenous retrovirus-K was also observed by the same authors in affected cortical neurons (CitationDouville & Nath, 2017). While an association between the two was inferred, the possibility that Tat, a neurotoxic HIV-1 protein detectable in the cerebrospinal fluid (CSF) of HIV-infected individuals (CitationHenderson et al., 2019; CitationJohnson et al., 2013), might contribute to the observed pathology was not tested. We investigated the possibility that Tat could be involved in TDP-43 overproduction or aggregation in the central nervous system (CNS) using an inducible Tat transgenic (Tg) mouse model. There are also factors unrelated to HIV that can worsen neuroHIV pathology. One such comorbid insult is opioid use disorder (OUD) (CitationFitting et al., 2010, Citation2020; CitationHauser et al., 2006; CitationMurphy et al., 2019), which can by itself dysregulate TDP-43 kinase (CitationChakrabarti et al., 2020; CitationWager et al., 2014). Hence, we also evaluated the impact of morphine, a prototypical opioid of abuse/misuse, in some outcome measures.
We focused on the basal ganglia, a major CNS target of HIV infection (CitationBerger & Arendt, 2000; CitationNewsome et al., 2011), for the human studies reported here. We further utilized a HIV-1 Tat Tg mouse model and primary striatal neuronal cultures to address the question of the possible contribution of Tat and/or morphine to the observed outcomes. Lastly, to help decipher the mechanism of the proteinopathy, we evaluated the levels and activity of two TDP-43 kinases (i.e., CK1δ and CK2) and the effects of antagonizing the kinases on the observed TDP-43 outcomes.
Increased cytoplasmic hyperphosphorylated TDP-43 was observed in HIV+ post-mortem brain tissues. Aberrant phosphorylation of TDP-43 coincided with increased CK2 immunoreactivity. Likewise, Tat expression in Tg mice increased cytoplasmic TDP-43 phosphorylation and CK2 levels, while CK1δ levels were unchanged in samples from post-mortem human basal ganglia or the mouse striatum following HIV or Tat exposure, respectively. Morphine increased cytoplasmic TDP-43 levels and augmented Tat-induced phosphorylation of TDP-43. CX-4945 (Silmitasertib), a US Food and Drug Administration (FDA) recently approved CK2 antagonist, attenuated the Tat- and morphine-induced pathologic phosphorylation of TDP-43 in primary mouse striatal neuronal cultures.
Materials and Methods
Post-Mortem HIV+ and HIV − Tissues
Post-Mortem Sample Collection and Subject Characteristics
A total of seven HIV+ (n = 7) and four seronegative (n = 4) post-mortem tissues were used in this study. All 4 of the seronegative tissues and 4 of the HIV+ tissues (formalin-fixed and paraffin-embedded posterior basal ganglia tissues) were obtained from the National NeuroAIDS Tissue Consortium. All patient data were coded, and tissues were handled per NIH guidelines to protect patient identities. Details of tissue collection and processing can be obtained from the National NeuroAIDS Tissue Consortium website (https://nntc.org/query/tool). Three additional tissue samples containing the tail of the caudate nucleus were obtained from post-mortems performed in Kampala, Uganda from 2017 to 2018 on a cohort of Ugandans with known HIV. Written informed consent was obtained from next of kin under a protocol approved by the Research Ethics Committee of Mulago National Referral Hospital. The tissues were collected within a median post-mortem interval of 4.7 h, snap-frozen, shipped to the United States, and stored at −80oC until use. Two (2) of the 7 HIV+ tissues were obtained from female subjects. Additional case-specific information on all human tissues used in the current study is detailed in .
Table 1 Subject Characteristics.
Immunofluorescence Assay
Paraffin-embedded tissues from the posterior basal ganglia (primarily, the caudate and putamen) of HIV+ and seronegative individuals were dewaxed and rehydrated in xylene (100%; 3 × 10 min), ethanol (100%; 2 × 10 min, 95%; 2 × 5 min, 70%; 2 × 5 min, 50%; 2 × 5 min), and dH2O (2 × 5 min) prior to immunofluorescence assay. Snap-frozen tissues were embedded in O.C.T compound, sectioned, and postfixed (with ice-cold 4% paraformaldehyde for 20 min) prior to use. All tissues (12–18 μm-thick) were heated (at 50% microwave power for 3 min) in Tris-based antigen unmasking solution (pH 9.0, # H3301, Vector Laboratories, Burlingame, CA), followed by permeabilization in a neutral pH phosphate-buffered saline (PBS) containing 0.25% Triton X 100. Tissue sections were incubated (2 h) in Animal-Free Blocker® and Diluent solution (# SP-5035-100, Vector Laboratories) and then incubated overnight (at 4oC) in primary antibodies for the 32 kDa dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP-32) (1:200, # sc271111 AF647, Santa Cruz, Dallas, TX), TDP-43 (1:100, # 67345, Proteintech, Rosemont, IL), phospho-TDP-43 Ser409/410 (pTDP-43) (1:100, # 66318, Proteintech), CK2 (1:100, #10992, Proteintech), and CK1δ (1:100, #14388, Proteintech) followed by a 1 h incubation in species-specific secondary antibodies. Autofluorescence in tissue sections was eliminated using ReadyProbes Tissue Autofluorescence Quenching Kit (#R37630, Thermo Fisher, Waltham, MA) per the manufacturer’s instructions. Mean fluorescence/pixel intensity values for pTDP-43, TDP-43, CK2, and CK1δ (corresponding to the level of immunostaining) were acquired in optical sections using confocal microscopy and measured in the cytoplasm and nuclear compartments using CellProfilerTM software (V 6.1) (Broad Institute, Cambridge, MA) (see supplementary for details on the CellProfilerTM workflow). At least 300 Hoechst+ cells were analyzed for each subject.
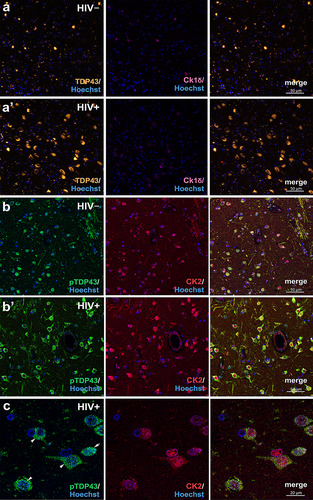
Figure 1 Pathologic deposition of TDP-43 and casein kinase 2 (CK2) immunoreactivity in the basal ganglia of HIV+ individuals. (a,a’ and b,b’) Representative confocal images of TDP-43, casein kinase 1δ (CK1δ), pTDP-43, and CK2 immunofluorescence in HIV− (a,b) and HIV+ (a’,b’) basal ganglia. Note the overall paucity of CK1δ antigenicity and the increased nuclear-to-cytoplasmic redistribution of TDP-43 antigenicity in the neural cells of HIV+ individuals. (c) A high magnification (63×) image showing pTDP-43 and CK2 immunofluorescence colocalization in cells within post-mortem HIV+ basal ganglia.
HIV-1 Tat Transgenic Mice
Animals
Twenty-four, male Tg mice (3–5 months old) expressing the HIV-1 tat (Tat+) or lacking the tat transgene (Tat−) were used for this study. Tg mice were generated and housed in an environmentally controlled vivarium (12 h light/dark cycle; lights off at 6 pm) at Virginia Commonwealth University. Food and water were provided ad libitum. Expression of Tat was targeted preferentially to the nervous system in Tg mice via a GFAP-driven, reverse tetracycline transactivator (rtTA or tetracycline-on promoter) that was activated with doxycycline (DOX)-containing mouse chow as previously described (CitationBruce-Keller et al., 2008; CitationHauser et al., 2009). Tat− mice expressing the rtTA promoter served as controls. DOX-containing chow (6 g/kg; Harlan Laboratories, Madison, WI) was freely available to both Tat+ and Tat− mice for 8 weeks. All procedures were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee and conformed to the Guide for Care and Use of Laboratory Animals (National Research Council, Washington DC).
Morphine Treatment
Morphine sulfate obtained from the National Institute on Drug Abuse Drug Supply Program, Bethesda, MD was dissolved in sterile physiological saline. Mice were administered a ramping morphine dose of 10 to 40 mg/kg subcutaneously, increasing by 10 mg/kg/day b.i.d. every second day (until 40 mg/kg/day b.i.d. was achieved on day 7 and continued thereafter) during the last 2 weeks of Tat induction (CitationNass et al., 2021, Citation2022; CitationOhene-Nyako et al., 2021). Control mice received an equivalent volume of saline.
Immunoblotting
Cytoplasmic and nuclear lysates were obtained from striatal samples using NE-PER nuclear and cytoplasmic extraction reagents (#78833, Thermo Fisher) per the manufacturer’s instructions. Briefly, striatal tissues were washed with neutral pH phosphate-buffered saline (PBS) and centrifuged at 500 × g for 5 min. After discarding the supernatant, samples were homogenized with a Dounce homogenizer in ice-cold CER I buffer, vortexed for 15 s, and incubated on ice for 10 min. A 1× solution of HALT protease and phosphatase inhibitor cocktail (Thermo Fisher) was added to all lysis reagents. Ice-cold CER II reagent was added to the homogenate, after which samples were vortexed and centrifuged at 16,000 × g for 5 min. The supernatant (containing the cytoplasmic fraction) was aliquoted and stored at −80oC. To obtain the nuclear fraction, the remaining pellet was resuspended in ice-cold NER solution, vortexed for 15 s every 10 min for a total of 40 min, then centrifuged at 16,000 × g for 10 min. Nuclear extracts were aliquoted and stored at −80oC. Protein concentration in lysates was determined by the bicinchoninic acid (BCA) method. 30 µg samples were electrophoresed on 10% Criterion TGX gels (Bio-Rad, Hercules, CA) and transferred to PVDF membranes. Non-specific binding sites on membranes were blocked for 1 h with Intercept blocking buffer (LI-COR, Lincoln, NE) for all kinase blots or 5% non-fat dry milk in Tris-buffered saline/Tween-20 (TBST) for pTDP-43/TDP-43 blots. Membranes were then incubated overnight in primary antibodies directed against pTDP-43 (Ser409/410) (1:1000, # 66318, Proteintech), CK2 (1:1,000, #10992, Proteintech), CK1δ (1:1,000, #14388, Proteintech), and inositol-triphosphate 3-kinase B (ITPKB) (1:1000, #12816, Proteintech) followed by incubation in species-specific HRP-conjugated secondary antibodies (for pTDP-43/TDP-43) or fluorescent-labeled secondary antibodies (for CK2, CK1δ, or ITPKB). For cytoplasmic extracts, GAPDH (# A303-878A; 1:2000; Bethyl Laboratories Inc., Montgomery, TX) was used to assess loading while LI-COR Total Protein Stain (# 92611011, LI-COR) was used to assess loading in nuclear extracts. Images of blots were obtained using a ChemiDoc Imager (Bio-Rad) and western blotting bands were analyzed with the Image Lab software (V6.0.1, Bio-Rad).
In Vitro Assays in Primary Striatal Neurons
Primary Striatal Neuronal Cultures and Drug Treatments
Primary striatal neurons were isolated from ICR mice at embryonic day 17 as previously described (CitationFitting et al., 2014; CitationZou et al., 2011). Briefly, striatal tissues were dissected, minced, and incubated for 30 min at 37oC in neurobasal medium (NBM) containing 0.015 mg/mL DNase and 2.5 mg/ml trypsin. NBM was supplemented with 25 mM glutamate, 0.5 mM glutamine, B27 (Invitrogen), and an antibiotic-antimycotic solution (Sigma). Cells were then triturated, filtered (2×) through a 70 μm-diameter pore membrane, and seeded into six-well plates (15 × 105 neurons/well) pre-coated with poly-L-lysine. Cells were maintained in NBM supplemented at decreasing concentrations of glutamate (days 1–4, 25 mM; days 5–6, 12.5 mM; days 7–11, 0 mM) at 37oC in a 5% CO2 environment until they matured (at 11 days). Based on our findings that TDP-43 phosphorylation was greatest following co-exposure to morphine and Tat in murine brain tissues, and to optimize detecting alterations in TDP-43 phosphorylation, we co-exposed cells to Tat and morphine to verify the involvement of CK2 in the pathologic TDP-43 phosphorylation. Cells were either treated with vehicle (DNase/RNase-free non-pyrogenic Ultrapure water), co-treated with Tat (100 nM) and morphine (500 nM), or a combination of Tat, morphine, and 0.5, 1, or 2 µM concentrations of the highly selective CK2 antagonist CX-4945 (#A11060, AdooQ Bioscience, Irvine, CA). CX-4945 (Silmitasertib) was granted Orphan Drug Designation by the US FDA in December 2021 for the treatment of medulloblastoma and potentially other rare diseases targeting CK2. Vehicle-treated cells served as controls. The selected concentrations of Tat and morphine do not affect cell viability within 24 h (CitationZou et al., 2011). The doses of CX-4945 selected for testing are those previously determined to reduce pro-inflammatory mediators to baseline levels in immune cells (CitationJang et al., 2017). Treated neurons were incubated at 37oC in a 5% CO2 environment for 24 h. Afterward, the cells were harvested, and the cytoplasmic and nuclear fractions were extracted using a NE-PER nuclear and cytoplasmic extraction kit (Thermo Fisher). BCA was used to determine protein concentrations and samples were stored in aliquots at −80oC until use.
Immunoblotting
Cytoplasmic lysates from the neuronal cultures (10 µg) were electrophoresed on 10% Criterion TGX gels (Bio-Rad) and transferred to PVDF membranes. Non-specific binding was blocked using 5% milk in TBST for 1 h at room temperature (RT). Membranes were incubated overnight at 4oC with primary antibodies for pTDP-43 (1:1000, # 66318, Proteintech) and TDP-43 (1:1000, # 67345, Proteintech). After washing in TBST, membranes were incubated in species-specific HRP-conjugated secondary antibodies for 1 h at RT. Membranes were scanned with a Bio-Rad ChemiDoc Imager and optical density was determined using Image Lab software (V6.0.1, Bio-Rad).
Enzyme Activity Assay
An enzyme activity assay was performed to determine whether Tat and morphine exposure alters CK2 enzyme kinetics. CK2 activity was determined in cytoplasmic extracts using the CycLex CK2 activity kit (# CY-1170, MBL Life Science, CycLex Co. Ltd, Nagano, Japan) per the manufacturer’s instructions and previous publications (CitationJung et al., 2014; CitationLee et al., 2014; CitationOinuma et al., 2010). Briefly, 5 µg samples obtained from lysates of cells co-treated with Tat and morphine, or vehicle-treated control striatal neurons (receiving no experimental treatments) were incubated in wells pre-coated with a synthetic CK2-specific peptide with a serine 46 moiety that can be phosphorylated by CK2. A peroxidase-coupled monoclonal antibody was used to selectively detect phosphorylation of the serine 46 moiety of CK2 via the conversion of tetramethylbenzidine (TMB) to a chromogenic reaction product. The TMB reaction product was read at an absorbance of 450 nm on a PHERAstar FS plate reader (BMG LabTech; Cary, NC). For this study, reaction/incubation times used were 0 min, 20 min, and 40 min. Due to the unavailability of a CK1 enzyme activity kit and since CK1 levels were not changed in HIV- or Tat-exposed tissues in the present study, further evaluation of CK1 activity did not seem warranted.
Quantification and Statistical Analyses
All outcomes from the HIV+ and HIV− human tissues were analyzed using Student’s t-test. The ratio of cytoplasmic to nuclear TDP-43 intensity was used as an index of protein mislocalization between the two compartments [e.g., (CitationBarmada et al., 2010)]. An estimation plot of the ratios was used to show the magnitude and precision of the calculated effect at a 95% confidence interval. Pearson correlation was used to determine the relationship between intensities of pTDP-43 and casein kinases.
For in vivo rodent experiments, data were standardized to Tat− mice exposed to saline. Data for immunoblots were analyzed using a two-way ANOVA followed by a post hoc Tukey multiple comparisons test. Pearson correlation was used to determine the relationship between pTDP-43 levels and casein kinase levels.
For in vitro experiments, data were standardized to vehicle-treated (control) striatal neurons. Data were analyzed using one-way ANOVA. A post hoc Tukey’s test was used for between-group differences. All statistical analyses were performed using GraphPad Prism v 9.0, (La Jolla, CA) with significance set at p < 0.05. Data are presented as the mean + the SEM.
Scientific Rigor
To reduce experimental bias and enhance rigor, all data were analyzed by a genotype-, case-, or treatment-blinded observer. A peptide blocking test was used to confirm pTDP-43 antibody specificity and to prevent analysis of non-target antigenic bands on immunoblots (pTDP-43 blocking peptide: #22309-1-BP, Proteintech). Other antibodies were validated using “orthogonal” or “independent antibody validation” tests (CitationUhlen et al., 2016). Specificity of CK2 activity assays was verified using a purified CK2 enzyme (#CY-E1170-1, Cyclex) and a CK2-specific antagonist (CX-4945).
Results
Post-Mortem HIV Human Tissues
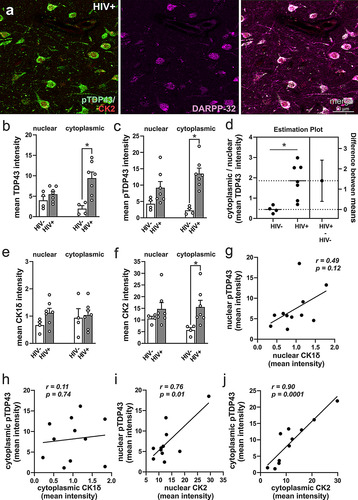
A previous study revealed increased TDP-43 levels in cortical neurons of HIV-infected individuals (CitationDouville & Nath, 2017). Here, we show the presence of the neurotoxic phosphorylated forms of TDP-43 (, b, and c), and pTDP-43 colocalization in DARPP-32+ basal ganglia neurons from post-mortem HIV+ brains () and in presumptive glia (see supplementary ). The intensity of nuclear TDP-43 immunostaining was similar in HIV+ and HIV− tissue samples (t = 0.78, two-tailed p = 0.46; ), while cytoplasmic TDP-43 was increased (t = 2.96, two-tailed p = 0.025; ) in HIV+ brain tissues compared to HIV− tissues. Likewise, no differences were observed for nuclear pTDP-43 intensity (t = 1.84, two-tailed p = 0.09; ) but cytoplasmic pTDP-43 was increased (t = 4.83, two-tailed p = 0.0009; ) in HIV+ compared to HIV− brain tissues. A ratio of cytoplasmic to nuclear TDP-43 also revealed aberrant localization (t = 5.27, two-tailed p = 0.002; ) of the protein in HIV+ brain tissues. Evaluation of the two kinases that phosphorylate TDP-43 (at Ser 409/410) revealed no changes to nuclear (t = 1.89, two-tailed p = 0.09; ) or cytoplasmic (t = 0.28, two-tailed p = 0.78; ) intensities of CK1δ. Although there were no differences in nuclear CK2 intensity (t = 1.06, two-tailed p = 0.33; ), cytoplasmic CK2 intensity was increased in HIV+ tissues (t = 2.49, two-tailed p = 0.03; ). Pearson correlation (pooled across both groups) revealed no relationship between nuclear pTDP-43 and CK1δ (r = 0.49, p = 0.12; g) or cytoplasmic pTDP-43 and CK1δ (r = 0.11, p = 0.74; h). On the contrary, a direct relationship was observed between nuclear pTDP-43 and CK2 (r = 0.78, p = 0.01; ), and cytoplasmic pTDP-43 and CK2 levels (r = 0.90, p < 0.0001; ).
Figure 2 Assessment of pathologic TDP-43 and casein kinase 2 (CK2) immunoreactivity in the basal ganglia of HIV− and HIV+ individuals. (a) Colocalization of pTDP-43, CK2, and DARPP32 immunoreactivity in cells of post-mortem HIV+ brains confirming the identity of the cells as medium spiny neurons. (b,c) Cytoplasmic TDP-43 (b) and pTDP-43 (c) fluorescence intensities were increased in HIV+ compared to HIV− basal ganglia. (d) Cytoplasmic-to-nuclear ratio of TDP-43 fluorescent intensity was increased in HIV+ tissues indicating mislocalization. (e) CK1δ fluorescent intensity was slightly above background levels and not changed in any of the groups evaluated. (f) Cytoplasmic CK2 intensity was increased in HIV+ compared to HIV− basal ganglia. Pearson correlations revealed no significant relationship between the nuclear (g) and cytoplasmic (h) intensities of pTDP-43 (pooled across groups) vs CK1δ fluorescence. On the contrary, nuclear (i) and cytoplasmic (j) intensities of pTDP-43 fluorescence (pooled across groups) correlated positively with CK2 fluorescence intensities. Mean intensity values indicate the mean fluorescence pixel intensities for each respective protein acquired in optical sections using confocal microscopy and analyzed using CellProfilerTM 6.1 software (see Materials and Methods 2.1.2). Data are expressed as the mean + the SEM (n = 4-7/group). * indicates a significant difference by Student’s t-test (p < 0.05).
HIV-1 Tat Transgenic Mice
We evaluated pTDP-43 and TDP-43 levels in the striatum of mice exposed to Tat and/or morphine. Two-way ANOVA revealed no significant changes in nuclear TDP-43 levels (Tat main effect: F(1,19) = 0.01, p = 0.94, morphine main effect: F(1,19) = 0.30, p = 0.59, interaction: F(1,19) = 1.98, p = 0.18; ). Likewise, no Tat main effect was observed for cytoplasmic TDP-43 levels (F(1,19) = 2.55, p = 0.13; ) but a significant main effect was observed for morphine to increase cytoplasmic TDP-43 levels (F(1,19) = 4.44, p = 0.048; ). No significant interaction (F(1,19) = 1.39, p = 0.25; ) or post hoc differences were observed. When nuclear pTDP-43 levels were evaluated in the same tissues, a main effect of Tat (F(1,20) = 0.27, p = 0.61; ) on nuclear pTDP-43 levels was not evident. By contrast, a significant morphine main effect (F(1,20) = 13.50, p = 0.002; ) was observed that did not interact with Tat (F(1,20) = 0.55, p = 0.47; ). Post hoc analyses revealed significant differences between morphine-treated Tat+ mice vs saline-treated Tat+ mice, and vs saline-treated Tat− mice. In the cytoplasm, main effects of Tat (F(1,20) = 9.05, p = 0.007; and c) and morphine (F(1,20) = 6.97, p = 0.02; and c) were observed for pTDP-43 levels, while there was no interaction between the two (F(1,20) = 0.03, p = 0.87; and c). Post hoc analysis revealed significant differences between morphine-treated Tat+ mice vs saline-treated Tat− mice. No differences were observed in the ratio of cytoplasmic to nuclear TDP-43 levels (Tat main effect: F(1,19) = 0.65, p = 0.43, morphine main effect: F(1,19) = 1.70, p = 0.21, interaction: F(1,19) = 1.86, p = 0.19; ). Corroborating the observations in the human studies, we show the presence of the TDP-43 and pTDP-43 in NeuN+ striatal neurons and in non-neuronal cells (including Iba-1+ microglia) in the brains of Tat Tg mice (see supplementary and c). We have also provided additional immunoblotting images of the outcomes presented here (see supplementary ).
Figure 3 Effects of morphine and Tat on the concentrations of TDP-43 and casein kinases in the mouse striatum. (a) Nuclear TDP-43 levels were not changed in any of the groups evaluated. (a,b) Alternatively, morphine exposure increased cytoplasmic TDP-43 levels (a), and nuclear and cytoplasmic levels of pTDP-43 (b), while Tat increased cytoplasmic pTDP-43 concentrations (b). (b) Post hoc comparisons showed differences in nuclear pTDP-43 between saline-treated (Tat+ and Tat−) mice vs mice co-exposed to Tat and morphine, and in cytoplasmic pTDP-43 levels between saline-treated Tat− mice vs mice co-exposed to Tat and morphine. (c) Representative immunoblots of cytoplasmic pTDP-43, TDP-43, CK1δ, CK2α, and CK2α’, and GAPDH. (d) The cytoplasmic-to-nuclear ratios of TDP-43 levels were not changed in any of the treatment groups. (e) Nuclear and cytoplasmic CK1δ levels were not changed in any of the groups evaluated. (f) When CK2 levels were evaluated, Tat increased nuclear CK2α levels, and either Tat or morphine alone increased cytoplasmic CK2α levels. (f) Post hoc comparisons of cytoplasmic CK2α levels showed differences between saline-treated Tat− mice vs Tat+ mice that were exposed to morphine. (g) Nuclear and cytoplasmic levels of the CK2α’ subunit were unaffected by Tat or morphine exposure. Data are expressed as the mean + the SEM (n = 5-6/group). # indicates significant main effects by two-way ANOVA; * indicates planned contrasts with post hoc Tukey’s test (p < 0.05).
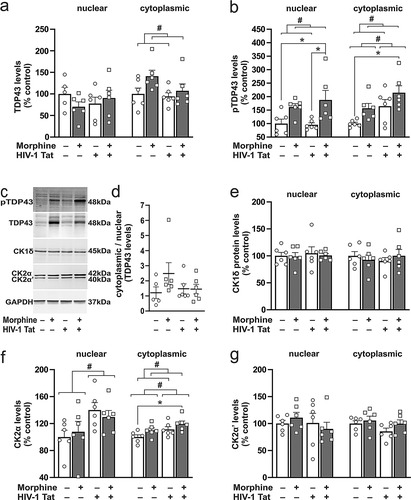
The finding that pTDP-43 levels were increased by Tat and morphine in the nuclear and cytoplasmic extracts led us to investigate the two kinases reported to phosphorylate TDP-43 at pathologic serine (Ser409/410) moieties. Similar to the observations in HIV+ brains, we did not observe any changes to nuclear (Tat main effect: F(1,20) = 0.18, p = 0.68, morphine main effect: F(1,20) = 0.11, p = 0.74, interaction: F(1,20) = 0.03, p = 0.87; ) and cytoplasmic (Tat main effect: F(1,20) = 0.01, p = 0.91, morphine main effect: F(1,20) = 0.01, p = 0.90, interaction: F(1,20) = 0.95, p = 0.34; ) levels of CK1δ. The antibody used for CK2 enabled the evaluation of the two catalytically active subunits of CK2 (i.e., CK2α and CK2α’). We observed a main effect for Tat (F(1,20) = 6.91, p = 0.02; ) to increase levels of nuclear CK2α, while a main effect (F(1,20) = 0.01, p = 0.91; ) or interaction (F(1,20) = 0.60, p = 0.45; ) with morphine was not observed. No significant differences were observed by post hoc comparisons. In the cytoplasm, levels of CK2α were increased by main effects of Tat (F(1,20) = 7.12, p = 0.01; ) and morphine (F(1,20) = 5.36, p = 0.03; ) with no interaction (F(1,20) = 0.04, p = 0.84; ) between the two. Post hoc analysis revealed significant differences between morphine-treated Tat+ mice vs saline-treated Tat− mice. The CK2α’ subunit was unchanged in either the nuclear (Tat main effect: F(1,20) = 0.65, p = 0.43, morphine main effect: F(1,20) < 0.01, p = 0.99, interaction: F(1,20) = 0.80, p = 0.38; g) or cytoplasmic extracts (Tat main effect: F(1,20) = 2.25, p = 0.15, morphine main effect: F(1,20) = 2.17, p = 0.16, interaction: F(1,20) = 0.43, p = 0.52; g) of any group evaluated.
To further understand the Tat- and/or morphine-mediated effects on pTDP-43 and CK2, we evaluated levels of ITPKB, the rate-limiting enzyme in the formation of inositol (1,3,4,5) tetrakisphosphate (IP4), a regulator of CK2 activity (CitationSolyakov et al., 2004). We observed a main effect for Tat to increase cytoplasmic ITPKB levels (F(1,20) = 6.52, p = 0.02). Although there was no main effect of morphine (F(1,20) = 0.44, p = 0.52), there was a marginally significant interaction between Tat and morphine to depress ITPKB levels (F(1,20) = 4.33, p = 0.05) (graph not shown). Post hoc analysis revealed a significant effect of Tat by itself to increase ITPKB.
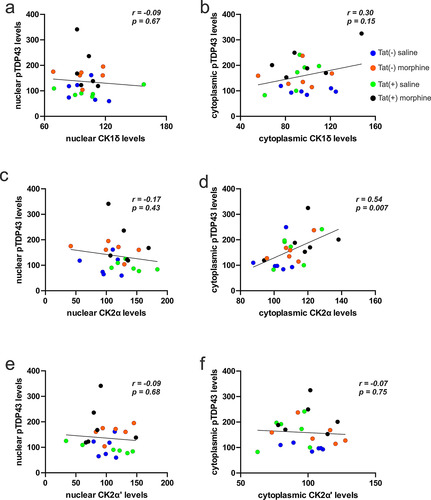
Levels of the individual TDP-43 kinases were correlated with pTDP-43 levels to determine the relationships between the changes in kinase levels and pTDP-43 accumulation. Pearson correlation (pooled across genotype and treatment) revealed no relationship between nuclear (r = -0.09, p = 0.67; ) and cytoplasmic (r = 0.30, p = 0.15; ) CK1δ levels vs pTD-P43 levels. Likewise, no significant correlation was observed between CK2α and nuclear pTDP-43 levels in the nucleus (r = -0.17, p = 0.43; ). However, a positive correlation was observed for cytoplasmic levels of CK2α and pTDP-43 (r = 0.54, p = 0.007; ), while no significant correlations were observed between nuclear (r = -0.09, p = 0.68; ) and cytoplasmic (r = -0.07, p = 0.75; ) CK2α’ levels vs pTDP-43 levels.
Figure 4 Effects of morphine and Tat on the relationship between the concentrations of pTDP-43 and TDP-43 kinases. (a,b) No significant correlations were observed between levels of nuclear (a) or cytoplasmic (b) CK1δ () and pTDP-43 (). (c) No significant correlations were observed between levels of nuclear CK2α () vs nuclear pTDP-43 (). (d) By contrast, levels of cytoplasmic CK2α () positively correlated with cytoplasmic pTDP-43 () concentrations (p < 0.05). (e,f) No significant correlations were observed between levels of nuclear (e) or cytoplasmic (f) CK2α’ () and pTDP-43 ().
In Vitro Assays in Primary Striatal Neurons
Primary striatal neurons provided a means to directly evaluate the effects of Tat and morphine on the cytoplasmic accumulation of pTDP-43 and to further verify the involvement of CK2. To determine the effects of CK2 inhibition on pTDP-43 levels, primary neurons co-exposed to Tat and morphine were treated concurrently with varying concentrations of the CK2 competitive antagonist CX-4945 (0.5, 1, and 2 µM) for 24 h. One-way ANOVA revealed no differences between group means for TDP-43 levels (F(4,16) = 0.14, p = 0.96; ). On the contrary, there were significant differences between group means for pTDP-43 levels as determined by one-way ANOVA (F(4,16) = 8.60, p = 0.0007; ). Post hoc analysis revealed that co-exposure to Tat and morphine increased pTDP-43 (p = 0.002; ). CX-4945 was able to prevent the Tat+/morphine effects on pTDP-43 accumulation, as planned contrasts showed significant differences in post hoc analyses when cells were treated with Tat and morphine only vs treatment with Tat and morphine and either 1 µM or 2 µM CX-4945 (p = 0.013 and p = 0.001, respectively; ).
Figure 5 The effects of Tat and morphine on pTDP-43 levels and CK2 activity in striatal neurons in vitro. (a) Tat or morphine exposure did not affect the concentration of cytoplasmic TDP-43. (b) By contrast, Tat and morphine co-exposure significantly increased levels of cytoplasmic pTDP-43, and these increases in pTDP-43 were fully reversible by the CK2 antagonist (CX-4945) in a concentration-dependent manner. (c) Tat and morphine co-exposure increased CK2 enzymatic activity in cytoplasmic extracts of striatal neurons. (d) Representative immunoblots of cytoplasmic pTDP-43, TDP-43, and GAPDH. Symbols indicate planned contrasts with post hoc Tukey’s test (p < 0.05). $ vs the control group (no active treatment), $$ vs the control group at the 20 min reaction time, $$$ vs the control group at 40 min, ǂ vs Tat+/morphine group, ^ vs Tat+/morphine and Cx-4945 at 20 min, ^^ vs Tat+/morphine and Cx-4945 at 40 min, * vs the 0 min reaction time. Data are expressed as the mean + or ± the SEM (n = 4 experiments/group).
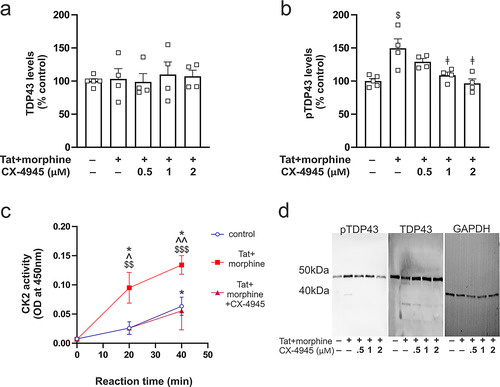
We further tested whether CK2 enzymatic activity was increased by Tat and morphine co-exposure. We observed significant main effects of reaction time (F(2,12) = 31.92, p < 0.0001) and treatment (F(2,12) = 21.09, p = 0.0006), and an interaction between the two (F(2,12) = 5.64, p = 0.02). Co-exposure to Tat and morphine increased CK2 activity after 20 min (p = 0.03; ) and 40 min (p = 0.02; ) when compared to control groups. Significant differences were also observed at 20 min (p = 0.007; ) and 40 min (p < 0.0001; ) following the onset (0 min) of the reaction with Tat and morphine co-exposure. On the contrary, only the 40 min reaction time showed a significant difference from baseline levels (0 min) in controls (p = 0.04; ). CX-4945 co-exposure prevented the Tat and morphine-induced increases in CK2 activity at 20 min (p = 0.02; ) and at 40 min (p = 0.01; ) of reaction time. Representative images of immunoblots are shown in .
Discussion
This study revealed the presence of pathologic pTDP-43 aggregates in the basal ganglia of post-mortem HIV-infected brain tissues and provides confirmatory in vivo and in vitro evidence that HIV-1 Tat and morphine increase the accretion of pathologic pTDP-43 in murine striatal neurons. Pathologic pTDP-43 deposition also appears to involve enhanced CK2 activity and protein levels.
Hyperphosphorylation of TDP-43 is a key feature of TDP-43 proteinopathy in several neurodegenerative disorders (CitationArai et al., 2010, Citation2009; CitationEck et al., 2021; CitationRen et al., 2021) and mediates pathologic aggregate formation. Here, we report increased pathologic pTDP-43 immunoreactivity in neurons of the posterior basal ganglia of post-mortem HIV+ brain. While the addition of post-mortem human tissues from HIV-infected individuals added value to the outcomes presented in the current study, we were unable to acquire certain relevant clinical and behavioral information on many of the subjects from whom the tissues were obtained. A limitation of our study is that some patient information including CNS viral titers, a prior history of substance abuse, antiretroviral therapy status, and the specific regions of the basal ganglia sampled are unavailable. The missing information might help better explain some of our observed outcomes and some of the within-group variances observed. The basal ganglia are particularly vulnerable to HIV infection and are sites where several neuropathological alterations have been reported (CitationBerger & Arendt, 2000; CitationDavison et al., 1997; CitationSanna et al., 2021; Citationvon Giesen et al., 2001). The pTDP-43 findings from the current study provide one explanation for this vulnerability. Tat-dependent increases in pTDP-43 in Tg mice corroborated observations from the human tissues in the current study and a previous report examining cortical tissues of HIV-infected individuals (CitationDouville & Nath, 2017). As Tat is detectable in the CSF of some HIV-infected individuals whose viral titers are controlled by cART (CitationJohnson et al., 2013; CitationHenderson et al., 2019), the observations from our Tat Tg mice suggest that hyperphosphorylated TDP-43 may persist even in some cART-treated individuals and may contribute to neurocognitive deficits in infected individuals. In both human and murine tissues, we observed TDP-43 antigenicity in neuronal and non-neuronal (glia-like) cell types (see Supplementary ). Investigating the implications of cell-specific and intracellular regulation of TDP-43 expression and activation is likely to provide considerable insight into the pathogenesis of neuroHIV. Cytoplasmic TDP-43 inclusions promote the formation of pathologic tau aggregates, interact with tau to enhance the deposition of insoluble complexes, and worsen neurodegenerative outcomes (CitationLatimer & Liachko, 2021; CitationLatimer et al., 2022; CitationMontalbano et al., 2020). We recently reported the presence of pathologic tau aggregates in striatal, amygdalar, and cerebral cortical brain regions with prolonged exposure to Tat and/or morphine (CitationKadri et al., 2015; CitationOhene-Nyako et al., 2021; CitationNass et al., 2022). Thus, the current findings provide a possible mechanism of HIV/HIV-1 Tat and opioid-induced neuronal injury via potential interactions between cytoplasmic pTDP-43 and pathologic tau aggregates. Such interactions have been observed in many neurodegenerative disorders and contribute to disease severity (reviewed in (CitationLatimer & Liachko, 2021)). Our results, therefore, may explain the apparent overlap in the pathobiology of neuroHIV and OUD.
In contrast to Tat, repeated administration of morphine dysregulated cytoplasmic TDP-43 levels suggesting that OUD may predispose to the development of TDP-43 proteinopathy. Although Tat and morphine independently increased cytoplasmic pTDP-43 levels, we did not observe any significant Tat and morphine interactions in the current study. The lack of an interaction may suggest that the effects of Tat were sufficiently robust to maximally dysregulate pTDP-43—masking any potential synergistic interactions that might be evident at lower morphine dosages or exposure durations. It is also possible that Tat and morphine might act via independent pathways to dysregulate pTDP-43. Despite the lack of Tat and morphine interactions, post hoc comparisons showed significant differences between Tat and morphine co-exposure and other treatment groups (e.g., ), suggesting that significant interactions may be apparent at different relative doses and/or longer exposure durations. Also, it is worth noting that some tissues used in the current study were obtained from individuals with a history of positive opioid drug tests. Whether prior/current opioid use contributed to the observed pTDP-43 pathology is unknown. However, the pathophysiologic alterations in pTDP-43 observed in morphine-exposed mice suggest this could be a possibility.
Importantly, we found that co-exposure to Tat and morphine altered CK2α levels. Although the two catalytic alpha subunits of CK2 exhibit functional redundancy, they are encoded by different genes (CitationPinna, 2002). Our findings suggest that the CK2α gene (CSNK2A1) is a target of Tat and morphine. It is also likely that Tat and morphine independently dysregulate the posttranslational modification, clearance, and/or cytoplasmic-to-nuclear shuttling of CK2α, which would explain their differential effects on pTDP-43 in the nucleus vs the cytoplasm. It can, however, be inferred from the CK2α outcomes that increases in enzyme levels alone may not determine pTDP-43 outcomes. Otherwise, Tat would increase nuclear pTDP-43 levels. Rather, additional factors that regulate the catalytic activity of the enzyme may also be important to the overall pTDP-43 outcomes. For example, IP4 can augment CK2 catalytic activity (CitationSolyakov et al., 2004). ITPKB, which converts inositol (1,4,5) triphosphate (IP3) to IP4 (CitationWen et al., 2004), was increased in the cytoplasm by Tat in the current study. Tat and morphine can also increase intracellular Ca2+ concentrations ([Ca2+]i) through both influx and mobilization from internal stores in striatal astroglia and neurons (CitationEl-Hage et al., 2005; CitationFitting et al., 2014; CitationHauser et al., 1996), which can stimulate IP3 generation through phospholipase C [e.g., (CitationChaudhry & Rubin, 1990; CitationMorrison & Shukla, 1988)]. It is, therefore, possible that ITPKB-dependent increases in the conversion of IP3 to IP4 in the cytoplasm may explain the differential regulation of TDP-43 phosphorylation in the nuclear and cytoplasmic compartments. This could also explain the increased CK2 activity in our in vitro assay. Other factors not evaluated in the current study, such as the activity of CK1δ, or other CK2-regulating inositol phosphates [e.g., IP5, IP6 (CitationSolyakov et al., 2004)], may contribute to the morphine effects on nuclear pTDP-43.
To conclude, pathological changes in neuronal and potentially glial pTDP-43 are a characteristic feature of neuroHIV and Tat expression may contribute to the aberrant phosphorylation of TDP-43 in mice. To the extent that results in Tat Tg mice can be generalized to humans, we speculate that elevations in pTDP-43 may persist even in cART-treated people living with HIV (PLWH) when CSF Tat is present. Our findings also suggest that OUD may contribute to TDP-43 proteinopathy. Targeting CK2 therapeutically might prove valuable in mitigating the abnormal deposition of neuronal pTDP-43 in PLWH or in people with OUD.
Author Contributions
MO-N and KFH conceptualized and designed the study. MO-N and SRN performed experiments and data acquisition. MO-N, SRN, KFH, PEK, wrote the original manuscript. MO-N, SRN, MPM, HTR, RL, MRN, KFH, PEK reviewed and edited the manuscript. RL, MRN, HTR, MPM, provided resources.
Availability of Data and Material
All data are included in this published article. Raw/unprocessed datasets are available from the corresponding author on reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
All tissues were collected and processed in accordance with the Declaration of Helsinki and per NIH ethical guidelines. For the tissues collected in Uganda, written informed consent was obtained from next of kin under a protocol approved by the Research Ethics Committee of Mulago National Referral Hospital, Kampala, Uganda.
Studies involving transgenic animals were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee and conformed to the Guide for Care and Use of Laboratory Animals (National Research Council, Washington DC).
ORCID iDs
Pamela E. Knapp https://orcid.org/0000-0003-4364-6074
Kurt F. Hauser https://orcid.org/0000-0001-7886-0332
sj-docx-1-asn-10.1177_17590914231158218 - Supplemental material for Casein Kinase 2 Mediates HIV- and Opioid-Induced Pathologic Phosphorylation of TAR DNA Binding Protein 43 in the Basal Ganglia
Download MS Word (1.4 MB)sj-docx-2-asn-10.1177_17590914231158218 - Supplemental material for Casein Kinase 2 Mediates HIV- and Opioid-Induced Pathologic Phosphorylation of TAR DNA Binding Protein 43 in the Basal Ganglia
Download MS Word (709.2 KB)sj-docx-3-asn-10.1177_17590914231158218 - Supplemental material for Casein Kinase 2 Mediates HIV- and Opioid-Induced Pathologic Phosphorylation of TAR DNA Binding Protein 43 in the Basal Ganglia
Download MS Word (948.5 KB)Acknowledgments
We greatly appreciate the support of the National Institute on Drug Abuse (NIDA) Drug Supply Program, which provided the opioids, and the National NeuroAIDS Tissue Consortium and the Mulago National Referral Hospital, Kampala, Uganda for providing postmortem CNS tissues used in these studies.
Supplemental Material
Supplemental Material
Supplemental Material
Funding
This publication was made possible by NIH funding from grants R01 DA045588, R01 DA034231, R01 DA057346, and F32 DA053163 (SRN) from NIDA; along with shared resources from NIH funding through the NIMH and NINDS by the following grants: Manhattan HIV Brain Bank (MHBB): U24MH100931, Texas NeuroAIDS Research Center (TNRC): U24MH100930, and the Data Coordinating Center (DCC): U24MH100925.
Supplemental material
Supplemental material for this article is available online.
Related Research Data
References
- Alquezar C., Salado I. G., de la Encarnación A., Pérez D. I., Moreno F., Gil C., de Munain A. L., Martínez A., Martín-Requero Á (2016). Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: A novel strategy for the treatment of frontotemporal dementia. Molecular Neurodegeneration, 11(1), 36. https://doi.org/10.1186/s13024-016-0102-7
- Arai T., Hasegawa M., Nonoka T., Kametani F., Yamashita M., Hosokawa M., Niizato K., Tsuchiya K., Kobayashi Z., Ikeda K., Yoshida M., Onaya M., Fujishiro H., Akiyama H. (2010). Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology: Official Journal of the Japanese Society of Neuropathology, 30(2), 170–181. https://doi.org/10.1111/j.1440-1789.2009.01089.x
- Arai T., Mackenzie I. R. A., Hasegawa M., Nonoka T., Niizato K., Tsuchiya K., Iritani S., Onaya M., Akiyama H. (2009). Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathologica, 117(2), 125–136. https://doi.org/10.1007/s00401-008-0480-1
- Barmada S. J., Skibinski G., Korb E., Rao E. J., Wu J. Y., Finkbeiner S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. Journal of Neuroscience, 30(2), 639–649. https://doi.org/10.1523/JNEUROSCI.4988-09.2010
- Berger J. R., Arendt G. (2000). HIV Dementia: The role of the basal ganglia and dopaminergic systems. J Psychopharmacol (Oxford), 14(3), 214–221. https://doi.org/10.1177/026988110001400304
- Bhargavan B., Woollard S. M., McMillan J. E., Kanmogne G. D. (2021). CCR5 Antagonist reduces HIV-induced amyloidogenesis, tau pathology, neurodegeneration, and blood-brain barrier alterations in HIV-infected hu-PBL-NSG mice. Molecular Neurodegeneration, 16(1), 78. https://doi.org/10.1186/s13024-021-00500-0
- Brady O. A., Meng P., Zheng Y., Mao Y., Hu F. (2011). Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. Journal of Neurochemistry, 116(2), 248–259. https://doi.org/10.1111/j.1471-4159.2010.07098.x
- Brown L. A. M., Scarola J., Smith A. J., Sanberg P. R., Tan J., Giunta B. (2014). The role of tau protein in HIV-associated neurocognitive disorders. Molecular Neurodegeneration, 9(1), 40. https://doi.org/10.1186/1750-1326-9-40
- Bruce-Keller A. J., Turchan-Cholewo J., Smart E. J., Geurin T., Chauhan A., Reid R., Xu R., Nath A., Knapp P. E., Hauser K. F. (2008). Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia, 56(13), 1414–1427. https://doi.org/10.1002/glia.20708
- Carlomagno Y., Zhang Y., Davis M., Lin W.-L., Cook C., Dunmore J., Tay W., Menkosky K., Cao X., Petrucelli L., Deture M. (2014). Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS One, 9(3), e90452. https://doi.org/10.1371/journal.pone.0090452
- Chakrabarti S., Liu N.-J., Gintzler A. R. (2020). Phosphorylation of unique C-terminal sites of the mu-opioid receptor variants 1B2 and 1C1 influences their Gs association following chronic morphine. Journal of Neurochemistry, 152(4), 449–467. https://doi.org/10.1111/jnc.14863
- Chaudhry A., Rubin R. P. (1990). Mediators of Ca2+-dependent secretion. Environmental Health Perspectives, 84, 35–39. https://doi.org/10.1289/ehp.908435
- Davison S. E., Aylward E. H., McArthur J. C., Selnes O. A., Lyketsos C., Barta P. E., Pearlson G. D. (1997). A quantitative MRI study of the basal ganglia in depression in HIV seropositive men. Journal of Neuro-Aids, 1(3), 29–41. https://doi.org/10.1300/J128v01n03_02
- Douville R. N., Nath A. (2017). Human endogenous retrovirus-K and TDP-43 expression bridges ALS and HIV neuropathology. Frontiers in Microbiology, 8, 1986. https://doi.org/10.3389/fmicb.2017.01986
- Eck R. J., Kraemer B. C., Liachko N. F. (2021). Regulation of TDP-43 phosphorylation in aging and disease. Geroscience, 43(4), 1605–1614. https://doi.org/10.1007/s11357-021-00383-5
- El-Hage N., Gurwell J. A., Singh I. N., Knapp P. E., Nath A., Hauser K. F. (2005). Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia, 50(2), 91–106. https://doi.org/10.1002/glia.20148
- Fitting S., Knapp P. E., Zou S., Marks W. D., Bowers M. S., Akbarali H. I., Hauser K. F. (2014). Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na+ influx, mitochondrial instability, and Ca2+ overload. Journal of Neuroscience, 34(38), 12850–12864. https://doi.org/10.1523/JNEUROSCI.5351-13.2014
- Fitting S., McRae M., Hauser K. F. (2020). Opioid and neuroHIV comorbidity - current and future perspectives. Journal of Neuroimmune Pharmacology: The Official Journal of the Society on NeuroImmune Pharmacology, 15(4), 584–627. https://doi.org/10.1007/s11481-020-09941-8
- Fitting S., Xu R., Bull C., Buch S. K., El-Hage N., Nath A., Knapp P. E., Hauser K. F. (2010). Interactive comorbidity between opioid drug abuse and HIV-1 Tat: Chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. American Journal of Pathology, 177(3), 1397–1410. https://doi.org/10.2353/ajpath.2010.090945
- Goossens J., Vanmechelen E., Trojanowski J. Q., Lee V. M. Y., Van Broeckhoven C., van der Zee J., Engelborghs S. (2015). TDP-43 as a possible biomarker for frontotemporal lobar degeneration: A systematic review of existing antibodies. Acta Neuropathologica Communications, 3(1), 15. https://doi.org/10.1186/s40478-015-0195-1
- Guerrero E. N., Mitra J., Wang H., Rangaswamy S., Hegde P. M., Basu P., Rao K. S., Hegde M. L. ( 2019a). Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Human Molecular Genetics, 28(15), 2459–2476. https://doi.org/10.1093/hmg/ddz062
- Guerrero E. N., Mitra J., Wang H., Rangaswamy S., Hegde P. M., Basu P., Rao K. S., Hegde M. L. ( 2019b). Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Human Molecular Genetics, 28(18), 3161–3162. https://doi.org/10.1093/hmg/ddz141
- Hasegawa M., Arai T., Nonaka T., Kametani F., Yoshida M., Hashizume Y., Beach T. G., Buratti E., Baralle F., Morita M., Nakano I., Oda T., Tsuchiya K., Akiyama H. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Annals of Neurology, 64(1), 60–70. https://doi.org/10.1002/ana.21425
- Hauser K. F., El-Hage N., Buch S., Nath A., Tyor W. R., Bruce-Keller A. J., Knapp P. E. (2006). Impact of opiate-HIV-1 interactions on neurotoxic signaling. Journal of Neuroimmune Pharmacology: The Official Journal of the Society on NeuroImmune Pharmacology, 1(1), 98–105. https://doi.org/10.1007/s11481-005-9000-4
- Hauser K. F., Hahn Y. K., Adjan V. V., Zou S., Buch S. K., Nath A., Bruce-Keller A. J., Knapp P. E. (2009). HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia, 57(2), 194–206. https://doi.org/10.1002/glia.20746
- Hauser K. F., Stiene-Martin A., Mattson M. P., Elde R. P., Ryan S. E., Godleske C. C. (1996). μ-Opioid receptor-induced Ca2+ mobilization and astroglial development: Morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca2+-dependent mechanism. Brain Research, 720(1-2), 191–203. https://doi.org/10.1016/0006-8993(96)00103-5
- Henderson L. J., Johnson T. P., Smith B. R., Reoma L. B., Santamaria U. A., Bachani M., Demarino C., Barclay R. A., Snow J., Sacktor N., Mcarthur J., Letendre S., Steiner J., Kashanchi F., Nath A. (2019). Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS (London, England), 33(Suppl 2), S145–S157. https://doi.org/10.1097/QAD.0000000000002268
- Huang W., Zhou Y., Tu L., Ba Z., Huang J., Huang N., Luo Y. (2020). TDP-43: From Alzheimer’s disease to limbic-predominant age-related TDP-43 encephalopathy. Frontiers in Molecular Neuroscience, 13, 26. https://doi.org/10.3389/fnmol.2020.00026
- Jang S. W., Hwang S. S., Kim H. S., Lee K. O., Kim M. K., Lee W., Kim K., Lee G. R. (2017). Casein kinase 2 is a critical determinant of the balance of Th17 and Treg cell differentiation. Experimental and Molecular Medicine, 49(9), e375. https://doi.org/10.1038/emm.2017.132
- Jiang L.-L., Xue W., Hong J.-Y., Zhang J.-T., Li M.-J., Yu S.-N., He J.-H., Hu H.-Y. (2017). The N-terminal dimerization is required for TDP-43 splicing activity. Scientific Reports, 7(1), 6196. https://doi.org/10.1038/s41598-017-06263-3
- Jiang L.-L., Zhao J., Yin X.-F., He W.-T., Yang H., Che M.-X., Hu H.-Y. (2016). Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Scientific Reports, 6(1), 23928. https://doi.org/10.1038/srep23928
- Jo M., Lee S., Jeon Y.-M., Kim S., Kwon Y., Kim H.-J. (2020). The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Experimental and Molecular Medicine, 52(10), 1652–1662. https://doi.org/10.1038/s12276-020-00513-7
- Johnson T. P., Patel K., Johnson K. R., Maric D., Calabresi P. A., Hasbun R., Nath A. (2013). Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proceedings of the National Academy of Sciences of the USA, 110(33), 13588–13593. https://doi.org/10.1073/pnas.1308673110
- Josephs K. A., Whitwell J. L., Weigand S. D., Murray M. E., Tosakulwong N., Liesinger A. M., Petrucelli L., Senjem M. L., Knopman D. S., Boeve B. F., Ivnik R. J., Smith G. E., Jack C. R., Parisi J. E., Petersen R. C., Dickson D. W. (2014). TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathologica, 127(6), 811–824. https://doi.org/10.1007/s00401-014-1269-z
- Jucker M., Walker L. C. (2018). Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nature Neuroscience, 21(10), 1341–1349. https://doi.org/10.1038/s41593-018-0238-6
- Jung E. B., Kim Y. J., Lee C. S. (2014). Casein kinase 2 inhibition attenuates cholesterol oxidation product-induced apoptosis by suppressing the activation of the mitochondrial pathway and the caspase-8- and bid-dependent pathways. Neurochemistry International, 65, 30–39. https://doi.org/10.1016/j.neuint.2013.12.010
- Kadri F., Pacifici M., Wilk A., Parker-Struckhoff A., Del Valle L., Hauser K. F., Knapp P. E., Parsons C., Jeansonne D., Lassak A., Peruzzi F. (2015). HIV-1-Tat protein inhibits SC35-mediated Tau Exon 10 inclusion through up-regulation of DYRK1A kinase. Journal of Biological Chemistry, 290(52), 30931–30946. https://doi.org/10.1074/jbc.M115.675751
- Latimer C. S., Liachko N. F. (2021). Tau and TDP-43 synergy: A novel therapeutic target for sporadic late-onset Alzheimer’s disease. Geroscience, 43(3), 1627–1634. https://doi.org/10.1007/s11357-021-00407-0
- Latimer C. S., Stair J. G., Hincks J. C., Currey H. N., Bird T. D., Keene C. D., Kraemer B. C., Liachko N. F. (2022). TDP-43 promotes tau accumulation and selective neurotoxicity in bigenic Caenorhabditis elegans. Disease Models & Mechanisms, 15(4), dmm049323. https://doi.org/10.1242/dmm.049323
- Lee H. R., Shin H. K., Park S. Y., Kim H. Y., Lee W. S., Rhim B. Y., Hong K. W., Kim C. D. (2014). Attenuation of β-amyloid-induced tauopathy via activation of CK2α/SIRT1: Targeting for cilostazol. Journal of Neuroscience Research, 92(2), 206–217. https://doi.org/10.1002/jnr.23310
- Montalbano M., McAllen S., Cascio F. L., Sengupta U., Garcia S., Bhatt N., Ellsworth A., Heidelman E. A., Johnson O. D., Doskocil S., Kayed R. (2020). TDP-43 and Tau oligomers in Alzheimer’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiology of Disease, 146, 105130. https://doi.org/10.1016/j.nbd.2020.105130
- Morrison W. J., Shukla S. D. (1988). Desensitization of receptor-coupled activation of phosphoinositide-specific phospholipase C in platelets: Evidence for distinct mechanisms for platelet-activating factor and thrombin. Molecular Pharmacology, 33(1), 58–63.
- Murphy A., Barbaro J., Martínez-Aguado P., Chilunda V., Jaureguiberry-Bravo M., Berman J. W. (2019). The effects of opioids on HIV neuropathogenesis. Frontiers in Immunology, 10, 2445. https://doi.org/10.3389/fimmu.2019.02445
- Nass S. R., Lark A. R. S., Hahn Y. K., McLane V. D., Ihrig T. M., Contois L., Napier T. C., Knapp P. E., Hauser K. F. (2021). HIV-1 Tat and morphine decrease murine inter-male social interactions and associated oxytocin levels in the prefrontal cortex, amygdala, and hypothalamic paraventricular nucleus. Hormones and Behavior, 133, 105008. https://doi.org/10.1016/j.yhbeh.2021.105008
- Nass S. R., Ohene-Nyako M., Hahn Y. K., Knapp P. E., Hauser K. F. (2022). Neurodegeneration within the amygdala is differentially induced by opioid and HIV-1 Tat exposure. Frontiers in Neuroscience, 16, 804774. https://doi.org/10.3389/fnins.2022.804774
- Nelson P. T., et al. (2019). Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain, 142(6), 1503–1527. https://doi.org/10.1093/brain/awz099
- Neumann M., Kwong L. K., Truax A. C., Vanmassenhove B., Kretzschmar H. A., Van Deerlin V. M., Clark C. M., Grossman M., Miller B. L., Trojanowski J. Q., Lee V. M.-Y. (2007). TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Journal of Neuropathology & Experimental Neurology, 66(3), 177–183. https://doi.org/10.1097/01.jnen.0000248554.45456.58
- Newsome S. D., Johnson E., Pardo C., McArthur J. C., Nath A. (2011). Fulminant encephalopathy with basal ganglia hyperintensities in HIV-infected drug users. Neurology, 76(9), 787–794. https://doi.org/10.1212/WNL.0b013e31820e7b4e
- Nonaka T., Suzuki G., Tanaka Y., Kametani F., Hirai S., Okado H., Miyashita T., Saitoe M., Akiyama H., Masai H., Hasegawa M. (2016). Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. Journal of Biological Chemistry, 291(11), 5473–5483. https://doi.org/10.1074/jbc.M115.695379
- Ohene-Nyako M., Nass S. R., Hahn Y. K., Knapp P. E., Hauser K. F. (2021). Morphine and HIV-1 Tat interact to cause region-specific hyperphosphorylation of tau in transgenic mice. Neuroscience Letters, 741, 135502. https://doi.org/10.1016/j.neulet.2020.135502
- Oinuma I., Ito Y., Katoh H., Negishi M. (2010). Semaphorin 4D/Plexin-B1 stimulates PTEN activity through R-Ras GTPase-activating protein activity, inducing growth cone collapse in hippocampal neurons. Journal of Biological Chemistry, 285(36), 28200–28209. https://doi.org/10.1074/jbc.M110.147546
- Ou S. H., Wu F., Harrich D., García-Martínez L. F., Gaynor R. B. (1995). Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. Journal of Virology, 69(6), 3584–3596. https://doi.org/10.1128/jvi.69.6.3584-3596.1995
- Peng C., Trojanowski J. Q., Lee V. M.-Y. (2020). Protein transmission in neurodegenerative disease. Nature Reviews. Neurology, 16(4), 199–212. https://doi.org/10.1038/s41582-020-0333-7
- Pinna L. A. (2002). Protein kinase CK2: A challenge to canons. Journal of Cell Science, 115(Pt 20), 3873–3878. https://doi.org/10.1242/jcs.00074
- Rayaprolu S., Fujioka S., Traynor S., Soto-Ortolaza A. I., Petrucelli L., Dickson D. W., Rademakers R., Boylan K. B., Graff-Radford N. R., Uitti R. J., Wszolek Z. K., Ross O. A. (2013). TARDBP Mutations in Parkinson’s disease. Parkinsonism & Related Disorders, 19(3), 312–315. https://doi.org/10.1016/j.parkreldis.2012.11.003
- Ren Y., Li S., Chen S., Sun X., Yang F., Wang H., Li M., Cui F., Huang X. (2021). TDP-43 and phosphorylated TDP-43 levels in paired plasma and CSF samples in amyotrophic lateral sclerosis. Frontiers in Neurology, 12, 663637. https://doi.org/10.3389/fneur.2021.663637
- Sanna P. P., Fu Y., Masliah E., Lefebvre C., Repunte-Canonigo V. (2021). Central nervous system (CNS) transcriptomic correlates of human immunodeficiency virus (HIV) brain RNA load in HIV-infected individuals. Scientific Reports, 11(1), 12176. https://doi.org/10.1038/s41598-021-88052-7
- Solyakov L., Cain K., Tracey B. M., Jukes R., Riley A. M., Potter B. V. L., Tobin A. B. (2004). Regulation of casein kinase-2 (CK2) activity by inositol phosphates. Journal of Biological Chemistry, 279(42), 43403–43410. https://doi.org/10.1074/jbc.M403239200
- Steinacker P., Barschke P., Otto M. (2019). Biomarkers for diseases with TDP-43 pathology. Molecular and Cellular Neuroscience, 97, 43–59. https://doi.org/10.1016/j.mcn.2018.10.003
- Tomé S. O., Vandenberghe R., Ospitalieri S., Van Schoor E., Tousseyn T., Otto M., von Arnim C. A. F., Thal D. R. (2020). Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathologica Communications, 8(1), 61. https://doi.org/10.1186/s40478-020-00934-5
- Tremblay C., St-Amour I., Schneider J., Bennett D. A., Calon F. (2011). Accumulation of transactive response DNA binding protein 43 in mild cognitive impairment and Alzheimer disease. Journal of Neuropathology & Experimental Neurology, 70(9), 788–798. https://doi.org/10.1097/NEN.0b013e31822c62cf
- Tsoi P. S., Choi K.-J., Leonard P. G., Sizovs A., Moosa M. M., MacKenzie K. R., Ferreon J. C., Ferreon A. C. M. (2017). The N-terminal domain of ALS-linked TDP-43 assembles without misfolding. Angewandte Chemie International Edition, 56(41), 12590–12593. https://doi.org/10.1002/anie.201706769
- Uhlen M., Bandrowski A., Carr S., Edwards A., Ellenberg J., Lundberg E., Rimm D. L., Rodriguez H., Hiltke T., Snyder M., Yamamoto T. (2016). A proposal for validation of antibodies. Nature Methods, 13(10), 823–827. https://doi.org/10.1038/nmeth.3995
- von Giesen H. J., Wittsack H. J., Wenserski F., Köller H., Hefter H., Arendt G. (2001). Basal ganglia metabolite abnormalities in minor motor disorders associated with human immunodeficiency virus type 1. Archives of Neurology, 58(8), 1281–1286. https://doi.org/10.1001/archneur.58.8.1281
- Wager T. T., et al. (2014). Casein kinase 1δ/ε inhibitor PF-5006739 attenuates opioid drug-seeking behavior. ACS Chemical Neuroscience, 5(12), 1253–1265. https://doi.org/10.1021/cn500201x
- Wang W., Arakawa H., Wang L., Okolo O., Siedlak S. L., Jiang Y., Gao J., Xie F., Petersen R. B., Wang X. (2017). Motor-Coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Molecular Therapy, 25(1), 127–139. https://doi.org/10.1016/j.ymthe.2016.10.013
- Wang W., Wang L., Lu J., Siedlak S. L., Fujioka H., Liang J., Jiang S., Ma X., Jiang Z., da Rocha E. L., Sheng M., Choi H., Lerou P. H., Li H., Wang X. (2016). The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nature Medicine, 22(8), 869–878. https://doi.org/10.1038/nm.4130
- Wen B. G., Pletcher M. T., Warashina M., Choe S. H., Ziaee N., Wiltshire T., Sauer K., Cooke M. P. (2004). Inositol (1,4,5) trisphosphate 3 kinase B controls positive selection of T cells and modulates Erk activity. Proceedings of the National Academy of Sciences of the USA, 101(15), 5604–5609. https://doi.org/10.1073/pnas.0306907101
- Wu C.-C., Jin L.-W., Wang I.-F., Wei W.-Y., Ho P.-C., Liu Y.-C., Tsai K.-J. (2020). HDAC1 Dysregulation induces aberrant cell cycle and DNA damage in progress of TDP-43 proteinopathies. EMBO Molecular Medicine, 12(6), e10622. https://doi.org/10.15252/emmm.201910622
- Zou S., Fitting S., Hahn Y.-K., Welch S. P., El-Hage N., Hauser K. F., Knapp P. E. (2011). Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at μ-opioid receptor-expressing glia. Brain, 134(Pt 12), 3616–3631. https://doi.org/10.1093/brain/awr281