Abstract
Extracellular superoxide dismutase (SOD3) is a secreted enzyme that regulates levels of extracellular superoxide and protects the extracellular matrix from degradation by reactive species. The SOD3 protein contains a heparin-binding domain and resides in a microenvironment rich in other heparin-bound growth factors, raising the possibility that SOD3 may have some biological role independent of its catalytic activity. To begin to address this, we designed and created enzymatically inactive mutant constructs targeting either the copper coordinating (i.e. H96 and H98) or superoxide channeling (i.e. N180 and R186) amino acid residues of SOD3. All constructs expressed equal quantities of immature intracellular SOD proteins, but only the N180A, R186A, and combination N180A/R186A mutants produced fully processed and secreted extracellular protein. Furthermore, while SOD activity was significantly inhibited in the single N180A and R186A mutants, the activity was completely abrogated in the N180A/R186A double mutant. Overall, the use of this novel tool may have broad reaching impacts into various fields of biology and medicine, and will aid in the delineation of cellular processes that are regulated by solely the SOD3 protein, its reactive oxygen species substrates and products, or the combination of both.
Introduction
Three distinct isoforms of the superoxide dismutase class of enzymes exist in humans (i.e. cytoplasmic (SOD1), mitochondrial (SOD2) and extracellular (SOD3)), and all have the unifying function of regulating superoxide flux in their respective subcellular compartments.Citation1 Utilizing copper and zinc (similar to the intracellular SOD1) in its active site, SOD3 catalyzes the reaction of superoxide to hydrogen peroxide at rates exponentially faster than spontaneous dismutation.Citation1 Due to the rapid reaction of superoxide with nitric oxide to create the highly reactive and damaging species peroxynitrite, it has been proposed that one of the major functions of SOD3 is to preserve nitric oxide bioavailability and limit peroxynitrite formation by the removal of superoxide.Citation2,Citation3 Indeed, in various experimental systems SOD3 has been demonstrated to protect vital components of the extracellular matrix (i.e. heparan sulfate) from degradation by peroxynitrite.Citation4–Citation6 Furthermore, unlike the ubiquitous expression of SOD1 and SOD2 in all tissues and cell types, SOD3 is only produced in a subset of cell types, which suggests a tissue-specific dependence upon the enzyme.Citation7 Due to this and other functions of SOD3, the study of this enzyme has broad-reaching impact potential in multiple areas of biology including cardiology, pulmonology, hepatology, hematology, and cancer.Citation5,Citation8–Citation12
Unlike the other superoxide dismutases, SOD3 possesses the unique feature of a heparin-binding domain.Citation13 This positively charged sequence of amino acids located at the C-terminal end of SOD3 allows for a tight association with the negatively charged heparan sulfate within the extracellular matrix, and additionally has been postulated to modulate SOD3 endocytosis.Citation14 This association with extracellular components is not an exclusive feature of SOD3, as many proteins possess heparin-binding domains, bind the extracellular matrix, and may become endocytosed in a similar manner.Citation15 Moreover, the vast majority of the proteins that bind heparan sulfate are factors involved in various biological processes such as coagulation, growth, inflammation, and immunity.Citation16 The juxtaposition of SOD3 with these various growth regulatory factors in the glycocalyx suggests that SOD3 may affect signaling functions beyond its catalytic activity.
In this study, we aimed to create a full-length catalytically inactive mutant of SOD3 in part to serve as a tool to query the enzyme-independent functions of this protein. To date, mutation studies of SOD3 have focused primarily on understanding the role of specific amino acids within the protein. For example, by systematic mutation of all cysteine residues in the enzyme, Jeon et al.Citation17 were able to identify critical disulfide linkages important in the proper folding and processing of SOD3. In addition, other studies have focused on various natural polymorphisms that may predispose a population to specific disease types.Citation8,Citation11,Citation18,Citation19 In these studies, the SOD3 variants examined were either improperly folded or still retained catalytic activity. We describe here for the first time a rationally targeted mutation to the active site of SOD3, which allows for a normally processed and excreted form of the enzyme retaining undetectable levels of catalytic activity. This new tool will enable the examination of non-catalytic effects SOD3 may possess, and serve as an important control in SOD3 over-expression studies to validate that the biological effects of SOD3 expression are due specifically to its catalytic activity.
Materials and methods
Vectors
The pcDNA3.1/Hygro(+) vector was purchased from Life Technologies, Carlsbad, CA, USA. Polymerase chain reaction (PCR) amplification of wild-type SOD3 was performed as previously described,Citation5,Citation12 and was cloned into pcDNA3.1/Hygro(+) using HindIII (5′) and XhoI (3′) restriction sites. Overlap extension PCR utilizing four different oligonucleotides was used for all mutant constructs.Citation20 Full-length extension oligonucleotides employed HindIII (5′) and XhoI(3′) restriction sites and were as follows: (5′)AAGCTTATGCTGGCGCTACTGTGTTCCTGC(3′) and (5′)CTCGAGTCAGGCGGCCTTGCACT(3′). Internal oligonucleotide sets for each mutant were as follows: H96A/H98A, (5′)CAGCCGCGCCATCGCCGTGGCCCAGTTCGGGACC(3′) and (5′)GGTCCCCGAACTGGGCCACGGCGATGGCGCGGCTG(3′); N180A, (5′)CAGGCCAGCGTGGAGGCCGGGAACGCGGGCC(3′) and (5′)GGCCCGCGTTCCCGGCCTCCACGCTGGCCTG(3′); R186A, (5′)GAAC GCGGGCCGGGCGCTGGCCTGCTGCG(3′) and (5′)CGCAGCAGGCCAGCGCCCGGCCCGCGTTC(3′). Sequential overlap extension PCR reactions were performed for N180A/R186A double mutant. PCR products were first inserted into the pJET1.2/blunt vector using the CloneJET PCR cloning system (Fermentas, Waltham, MA, USA), and subsequently subcloned into pcDNA3.1/Hygro(+) exploiting a HindIII and XhoI restriction digest. All vectors were validated by sequencing at the University of Iowa DNA Facility.
Cells and transfection
Human embryonic kidney cells (HEK293) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), and were used for all experiments. HEK293 cells were chosen due to their high transfection efficiency as well as the absence of endogenous SOD3 production. Cells were maintained in phenol red-free Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS). Transfection of plasmid DNA was carried out using Lipofectamine 2000 (Life Technologies) as per the manufacturer's instructions.
Quantitative real-time reverse transcriptase-PCR
Total RNA was extracted using the Trizol method and was quantified by the use of a Nanodrop ND-1000. One microgram from each sample was reverse transcribed using the Applied Biosystems cDNA archive kit (Applied Biosystems, Carlsbad, CA, USA). Generated cDNA was then subjected to SYBR green quantitative real-time PCR with primers specific to the coding sequence of SOD3 ((5′)GATCCGAGACATGTACGCCAA(3′) and (5′)TGCATGACCTCCTGCCAGA(3′)) and the 18 seconds control ((5′)GCCCGAAGCGTTTACTTTGA(3′) and (5′)TCATGGCCTCAGTTCCGAA(3′)) under the following PCR parameters: 95°C for 10 minutes, followed by 35 cycles of 95°C for 15 seconds, and 60°C for 1 minutes. A threshold in the linear range of PCR amplification was selected and the cycle threshold (Ct) determined. Levels of transcripts were then normalized to the 18 seconds control and compared relative to the untransfected control using the ΔΔCt method.
Western blotting
For extracellular protein analysis, transfections were performed using a minimal volume of media supplemented with 1% FBS. At time of analysis, media was harvested and cells were removed using a low protein binding 0.45 µm syringe filter. For intracellular protein analysis, protein was extracted using standard RIPA buffer and was quantified on the basis of the Bradford assay and a standard curve. Protein was run on an SDS–PAGE gel for separation of different-sized proteins followed by transfer to a nitrocellulose membrane. Identification and quantification of the amount of protein were performed by addition of specific primary antibody and then the addition of a horseradish peroxidase-tagged secondary antibody. Chemiluminescent substrate (ECL plus, GE Healthcare Life Sciences, Pittsburgh, PA, USA) was added to the membrane, film exposed, and then visualized bands were qualitatively quantified for protein amount. Loading errors were controlled for by normalizing to β-actin. Antibodies used for this study were SOD321 and β-tubulin (University of Iowa Hybridoma Core).
Electron paramagenetic resonance and SOD3 activity
For all experiments, conditioned media from HEK293 cells transfected for 48 hours with the appropriate SOD3 constructs were used. For electron paramagenetic resonance (EPR), conditioned media were incubated with 500 µmol/l hypoxanthine (Sigma-Aldrich, St. Louis, MO, USA) and 40 units/ml xanthine oxidase (Sigma, USA) in the presence of 50 mmol/l DMPO spin trap. The samples were analyzed using an AquaX (4-bore) sample cell and a Bruker EMX spectrometer (Bruker Biospin, Fremont, CA, USA). Signal heights were measured using the second downfield peak of the four-line DMPO spin adduct and reported in arbitrary amplitude units. The SOD3 spectrophotometric activity assay was performed on conditioned media using the WST-1 SOD assay kit (Dojindo Molecular Technologies, Rockville, MD, USA) as per the manufacturer's instructions.Citation22 Gel-based SOD3 activity assay was performed as previously described.Citation5,Citation12,Citation23 Briefly, after separation of 50 µl of conditioned cell culture media on a non-denaturing native gel using electrophoresis, the gel was stained with 2.4 mM nitro blue tetrazolium, 28 µM riboflavin, and 28 mM N,N,N,N-tetramethylethylenediamine for 20 minutes in darkness. SOD3 activity was visualized by illumination of the gel under a fluorescent light until achromatic bands appeared.
Statistics
Data are expressed as means and standard deviation. All experiments were performed on three separate biological replicates. Comparisons between groups were analyzed by the unpaired two-tailed Student's t-test. A P value ≤0.01 was considered significant.
Results
Rational design of a catalytically inactive SOD3
A variety of mutant constructs have been created to elucidate the function of specific amino acid residues or sequences in SOD3.Citation8,Citation11,Citation17–Citation19 While these rational and natural mutations have illuminated novel aspects of SOD3 in various physiologies and pathologies, data are limited regarding the role, if any, of the SOD3 protein in the absence of catalytic activity. Thus, we aimed in this study to rationally create a mature SOD3 protein that was deficient in its ability to dismute superoxide.
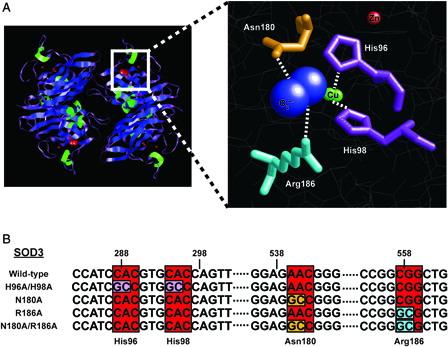
Examination of the crystal structure of human SOD3 revealed specific amino acid residues critical in the coordination of the active site of the enzyme.Citation24 Two histidine residues (96 and 98) participate in the coordination of the copper molecules, while an asparagine (180) and arginine (186) have been proposed stabilize the superoxide molecule and facilitate its passage to the copper catalytic site (A). For the creation of the SOD3 mutants, each of the aforementioned amino acids were mutated (either alone or in combination) using site-directed mutagenesis to alanine to eliminate any coordination contributions, and potentially render the protein inactive (B).
Figure 1. Rational design of a catalytically inactive SOD3 protein. (A) Left, cartoon schematic depicting tetrameric structure of SOD3 (Adapted from PDB ID: 2JLP). Blue = beta sheets; green = alpha helicies; purple = linker regions; red = all non-peptide components. Right, cartoon representation and magnification of active site. Residues targeted for mutation are indicated. Red ball = zinc atom; green balls = copper atoms; blue balls = superoxide molecule. (B) Sequence identification and validation of point mutations for each SOD3 mutant construct. Red boxes indicate DNA codons, while other colors correspond to respective amino acid residue in part (A).
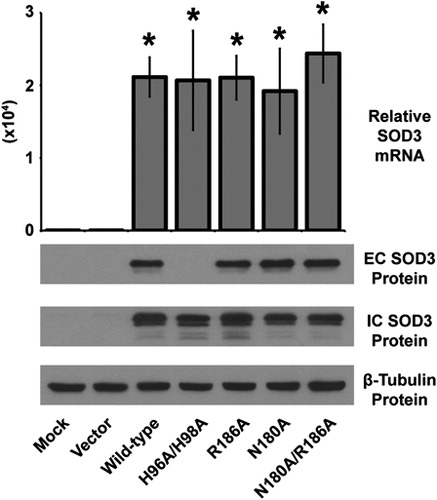
SOD3 mutants demonstrate variable extracellular expression
Because the mutations described here are of a rational design (as opposed to natural), it was prudent to characterize expression levels of each construct. Quantitative real-time reverse transcriptase (RT)-PCR analysis of SOD3 mRNA demonstrated an abundant and significant over-expression of each construct in HEK293 cells that do not express endogenous SOD3 (). In addition, intracellular (most likely pre-processed peptide from the endoplasmic reticulum) protein of all the mutant constructs was also highly expressed, and comparable to levels of wild-type SOD3 (). In contrast, only the H96A/H98A mutant revealed an absence in extracellular protein suggesting these histidine residues are critical to the overall structure and/or folding of the protein, and thus loss of these amino acids render the protein unable to become excreted (). Overall, the mutation of asparagine 180 and arginine 186 (alone or in combination) to alanine appears to have minimal effect on protein expression, stability, or excretion making these mutants optimal candidates for further analysis.
Figure 2. Characterization of the over-expression of the SOD3 mutant constructs. Upper, quantitative real-time RT-PCR analysis of RNA extracted from HEK293 cells transfected with SOD3 mutant constructs for 48 hours. Data normalized to 18 seconds loading control, then to mock transfected by ΔΔCT method. Data represent three biological replicates and are shown as mean and s.d. Where applicable, *P < 0.01 by Student's t-test versus mock-transfected. Lower, western blot analysis of extracellular (EC – from media) or intracellular (IC – from cells) SOD3 protein from HEK293 cells transfected with SOD3 mutant constructs for 48 hours. β-Tubulin shown for loading control.
The N180A/R186A mutant possesses undetectable levels of catalytic activity
While the N180A, R186A, and N180A/R186A mutants produced viable protein, the question still remained as to the catalytic activity of each construct. Qualitative SOD3 activity assay gels demonstrated that both the N180A and R186A single mutants still retained some activity, though it was strongly diminished (A). In contrast, the N180A/R186A mutant showed no detectable activity by the gel-based assay (B), which prompted further quantitative verification of catalytic activity.
Figure 3. The N180A/R186A mutant SOD3 demonstrates undetectable catalytic activity. (A and B) NBT reduction native gel electrophoresis assay for SOD3 on conditioned media from HEK293 cells transfected with SOD3 mutant constructs for 48 hours. (C) EPR spectra of the DMPO-OH adduct in the presence of the xanthine/xanthine oxidase superoxide-generating system. Each spectrum represents the system in the presence of conditioned media from HEK293 cells transfected with the respective SOD3 construct for 48 hours. All spectra are normalized to the same scale, and amplitude recorded in arbitrary units. (D) Quantification of a WST-1-based spectrophotometric SOD activity assay performed on conditioned media from HEK293 cells transfected with the respective SOD3 construct for 48 hours. Data demonstrate relative inhibition of xanthine/xanthine oxidase superoxide production. Data represent three biological replicates and are shown as mean and s.d. Where applicable, *P < 0.01 by Student's t-test versus vector-transfected.

Catalytic activity was first measured on the basis of EPR. Using an exogenous superoxide generating system (i.e. xanthine/xanthine oxidase), it was determined that while the wild-type SOD3 significantly attenuated the superoxide signal, the N180A/R186A mutant was not capable of eliminating superoxide with any greater efficiency than the vector control (C). Furthermore, to validate these findings a spectrophotometric assay based on 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-1) reduction by superoxide was performed.Citation22 While the wild-type SOD3 almost completely inhibited the reaction, the N180A/R186A mutant demonstrated no significant inhibition from the vector control (D). These data suggest that the N180A/R186A SOD3 mutant is a full-length excreted form of SOD3 that possesses undetectable levels of catalytic activity.
Discussion
The importance of SOD3 in various biological systems has been described both in vitro and in vivo. To date, SOD3 has only demonstrated a role as an extracellular antioxidant, but recent research has shown SOD3 translocation into the cytoplasm.Citation14,Citation25 This heparan sulfate-mediated endocytosis allows for the relocation of this antioxidant enzyme in close proximity with many potential intracellular signaling molecules. While the catalytic activity of SOD3 may initiate a signaling cascade, the mere presence of the SOD3 protein inside of the cell may also have a significant effect on cellular processes. The catalytically deficient mutant created here will serve as a valuable tool in elucidating the nature of SOD3 in intracellular signaling.
The SOD3 protein is processed through the endoplasmic reticulum–Golgi pathway where it becomes loaded with copper by way of the copper chaperone Antioxidant-1 (Atox1), glycosylated, and excreted to interact with the extracellular matrix.Citation26–Citation28 Due to Atox1 being strictly cytosolic, the copper is transported into the trans-Golgi network by the Menkes ATPase (ATP7A) where it is then loaded into the SOD3 enzyme prior to secretion.Citation29,Citation30 Once the enzyme is fully mature and extracellularly located, only then may it reenter the cell by endocytosis and mediate intracellular events.Citation14,Citation25 In this report we show that the N180A/R186A mutant of SOD3 is an excreted form of the enzyme that retains no catalytic activity. Interestingly, we also demonstrate that the copper-coordination mutant H96A/H98A is made in abundance intracellularly, but is not excreted. We hypothesize that this lack of copper coordination creates an improperly folded protein that is degraded prior to secretion. Jeon et al.Citation17 showed similar decreases in secreted SOD3 when creating systematic cysteine mutations necessary for disulfide linkages,Citation17 which suggests that higher-order structure of the protein is necessary for proper extracellular relocation. The N180A/R186A mutant does not perturb structurally relevant amino acids, and as such appears to be processed in a similar manner to wild-type SOD3.
Since the discovery of the superoxide dismutases by McCord and Fridovich,Citation31 their accepted role as purely antioxidant enzymes has remained static. Recently, many enzymes have been discovered to possess multiple functions, and we speculate that SOD3 may also possess activities other than superoxide dismutation. One of the first described examples of an enzyme that possessed a function other than its catalytic activity was the kinase Pbs2p in yeast. While Pbs2p was a long established MAPK kinase, it was discovered to also act as a cellular scaffold protein.Citation32 A more recent example is the hypoxia-inducible factor (HIF) prolyl hydroxylase 3 (PHD3). Normally, PHD3 acts to hydroxylate HIF, which targets the transcription factor for degradation during normoxic conditions. Recently, it has been reported that PHD3 is able to disrupt pyruvate kinase function in a non-catalytic manner, which leads to alterations in metabolism and cellular proliferation.Citation33,Citation34 In fact, over the last decade a substantial number of enzymes have demonstrated functions that are independent of their known catalytic activity.Citation35 While SOD3 is not currently described to possess biological activities other than the dismutation of superoxide, the mutant version described here will allow for further examination into other potential functions of this enzyme.
Thus far, research on superoxide dismutases has primarily focused on their inherent catalytic activity. Unlike the other superoxide dismutases, the structural similarity of extracellular superoxide dismutase with other extracellular signaling molecules suggests the potential for functions other than catalysis. Here, we describe for the first time the rational design and implementation of full-length and secreted mutant of extracellular superoxide dismutase for the elucidation of any non-catalytic dependent mechanisms of this enzyme. While the studies performed here utilized transient transfection in an in vitro system, the N180A/R186A mutant could be applied to in vivo systems through various viral constructs such as adenovirus, retrovirus, or adeno-associated virus. While this new tool will aid in the identification of new functions of SOD3, it will also assist the understanding of ROS-dependent processes in biology making it a valuable device in the arsenal of molecular biology-based free radical research.
Author Disclosure Statement
The authors declare no financial, personal, or professional competing interest with regard to this work.
Acknowledgements
This work was supported by NIH grant CA115438 (FED). We thank the University of Iowa DNA facility for their help in plasmid sequencing. We also thank Dr Garry Buettner and Brett Wagner and the Radiation and Free Radical Research Core of the Holden Comprehensive Cancer Center supported by NIH P30 CA086862 for assistance with the EPR studies. J.E.H. and J.J.M. received salary support from T32CA148062. Finally, we thank Dr Apollina Goel for the use of a Tecan spectrophotometer.
References
- Fridovich I. Superoxide dismutases. Annu Rev Biochem. 1975;44:147–59. Epub 1975/01/01.
- Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 2011;15(6):1583–606. Epub 2011/04/09.
- Gao F, Kinnula VL, Myllarniemi M, Oury TD. Extracellular superoxide dismutase in pulmonary fibrosis. Antioxid Redox Signal 2008;10(2):343–54. Epub 2007/11/15.
- Kliment CR, Tobolewski JM, Manni ML, Tan RJ, Enghild J, Oury TD. Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxid Redox Signal 2008;10(2):261–8. Epub 2007/10/27.
- Teoh ML, Fitzgerald MP, Oberley LW, Domann FE. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res 2009;69(15):6355–63. Epub 2009/07/16.
- Yao H, Arunachalam G, Hwang JW, Chung S, Sundar IK, Kinnula VL, et al. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci USA 2010;107(35):15571–6. Epub 2010/08/18.
- Nozik-Grayck E, Suliman HB, Piantadosi CA. Extracellular superoxide dismutase. Int J Biochem Cell Biol 2005;37(12):2466–71. Epub 2005/08/10.
- Arcaroli JJ, Hokanson JE, Abraham E, Geraci M, Murphy JR, Bowler RP, et al. Extracellular superoxide dismutase haplotypes are associated with acute lung injury and mortality. Am J Respir Crit Care Med 2009;179(2):105–12. Epub 2008/10/25.
- Break TJ, Jun S, Indramohan M, Carr KD, Sieve AN, Dory L, et al. Extracellular superoxide dismutase inhibits innate immune responses and clearance of an intracellular bacterial infection. J Immunol 2012;188(7):3342–50. Epub 2012/03/07.
- Iida S, Chu Y, Weiss RM, Kang YM, Faraci FM, Heistad DD. Vascular effects of a common gene variant of extracellular superoxide dismutase in heart failure. Am J Physiol Heart Circ Physiol 2006;291(2):H914–20. Epub 2006/07/15.
- Tamai M, Furuta H, Kawashima H, Doi A, Hamanishi T, Shimomura H, et al. Extracellular superoxide dismutase gene polymorphism is associated with insulin resistance and the susceptibility to type 2 diabetes. Diabetes Res Clin Pract 2006;71(2):140–5. Epub 2005/07/02.
- Teoh ML, Sun W, Smith BJ, Oberley LW, Cullen JJ. Modulation of reactive oxygen species in pancreatic cancer. Clin Cancer Res 2007;13(24):7441–50. Epub 2007/12/21.
- Adachi T, Kodera T, Ohta H, Hayashi K, Hirano K. The heparin binding site of human extracellular-superoxide dismutase. Arch Biochem Biophys 1992;297(1):155–61. Epub 1992/08/15.
- Chu Y, Piper R, Richardson S, Watanabe Y, Patel P, Heistad DD. Endocytosis of extracellular superoxide dismutase into endothelial cells: role of the heparin-binding domain. Arterioscler Thromb Vasc Biol 2006;26(9):1985–90. Epub 2006/07/01.
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 1999;68:729–77. Epub 2000/06/29.
- Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl 2002;41(3):391–412. Epub 2002/12/20.
- Jeon B, Kim BH, Lee YS, Kim S, Yoon JB, Kim TY. Inactive extracellular superoxide dismutase disrupts secretion and function of active extracellular superoxide dismutase. BMB Rep 2011;44(1):40–5. Epub 2011/01/27.
- Chu Y, Alwahdani A, Iida S, Lund DD, Faraci FM, Heistad DD. Vascular effects of the human extracellular superoxide dismutase R213G variant. Circulation 2005;112(7):1047–53. Epub 2005/08/10.
- Sorheim IC, DeMeo DL, Washko G, Litonjua A, Sparrow D, Bowler R, et al. Polymorphisms in the superoxide dismutase-3 gene are associated with emphysema in COPD. COPD 2010;7(4):262–8. Epub 2010/08/03.
- Reikofski J, Tao BY. Polymerase chain reaction (PCR) techniques for site-directed mutagenesis. Biotechnol Adv 1992;10(4):535–47. Epub 1992/01/01.
- Oury TD, Chang LY, Marklund SL, Day BJ, Crapo JD. Immunocytochemical localization of extracellular superoxide dismutase in human lung. Lab Investig 1994;70(6):889–98. Epub 1994/06/01.
- Ukeda H, Shimamura T, Tsubouchi M, Harada Y, Nakai Y, Sawamura M. Spectrophotometric assay of superoxide anion formed in Maillard reaction based on highly water-soluble tetrazolium salt. Anal Sci 2002;18(10):1151–4. Epub 2002/10/29.
- Teoh-Fitzgerald ML, Fitzgerald MP, Jensen TJ, Futscher BW, Domann FE. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis. Mol Cancer Res 2012;10(1):40–51. Epub 2011/11/09.
- Antonyuk SV, Strange RW, Marklund SL, Hasnain SS. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 A resolution: insights into heparin and collagen binding. J Mol Biol 2009;388(2):310–26. Epub 2009/03/18.
- Ohta H, Adachi T, Hirano K. Internalization of human extracellular-superoxide dismutase by bovine aortic endothelial cells. Free Radic Biol Med 1994;16(4):501–7. Epub 1994/04/01.
- Fukai T, Folz RJ, Landmesser U, Harrison DG. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res 2002;55(2):239–49. Epub 2002/07/19.
- Jeney V, Itoh S, Wendt M, Gradek Q, Ushio-Fukai M, Harrison DG, et al. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ Res 2005;96(7):723–9. Epub 2005/03/12.
- Petersen SV, Oury TD, Ostergaard L, Valnickova Z, Wegrzyn J, Thogersen IB, et al. Extracellular superoxide dismutase (EC-SOD) binds to type i collagen and protects against oxidative fragmentation. J Biol Chem 2004;279(14):13705–10. Epub 2004/01/23.
- Qin Z, Gongora MC, Ozumi K, Itoh S, Akram K, Ushio-Fukai M, et al. Role of Menkes ATPase in angiotensin II-induced hypertension: a key modulator for extracellular superoxide dismutase function. Hypertension 2008;52(5):945–51. Epub 2008/09/05.
- Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J 2006;20(2):334–6. Epub 2005/12/24.
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969;244(22):6049–55.
- Posas F, Saito H. Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 1997;276(5319):1702–5. Epub 1997/06/13.
- Chen N, Rinner O, Czernik D, Nytko KJ, Zheng D, Stiehl DP, et al. The oxygen sensor PHD3 limits glycolysis under hypoxia via direct binding to pyruvate kinase. Cell Res 2011;21(6):983–6. Epub 2011/04/13.
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011;145(5):732–44. Epub 2011/05/31.
- Rauch J, Volinsky N, Romano D, Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun Signal 2011;9(1):23–0. Epub 2011/11/01.