
ABSTRACT
Introduction. Despite many clinical efforts, non-small-cell lung cancer (NSCLC) has a dismal 5-year survival rate of 16%, and high incidence of recurrence. The success of biologically targeted agents, as well as the activity of well-established chemotherapeutic regimens, has been limited by inherited/acquired resistance, and biomarkers to adapt the prescription of anticancer drugs to patients’ features are urgently warranted.
Areas covered. In oncology, pharmacogenetics should provide the way to select patients who may benefit from a specific therapy that best match the individual and tumor genetic profile, thus allowing maximum activity and minimal toxicity. The present review summarizes the main findings on NSCLC pharmacogenetics, critically reappraising the most important studies on polymorphisms correlated with outcome of pemetrexed and EGFR-inhibitors, and provides perspective on clinical application of genomic tests for treatment decision-making.
Expert Opinion. A major challenge in NSCLC is the identification of subgroups of diseases/patients that will truly benefit from specific treatments. Ideally, convenient and minimally invasive tests to decipher biomarkers of chemosensitivity/resistance and toxicity should be developed alongside novel anticancer treatments. Integration with the latest generation of whole-genome analyses and liquid biopsies as well as prospective validation in large cohorts of patients will overcome the limitations of the traditional pharmacogenetic approaches.
1. Introduction
Parameters such as disease stage and grade, age, performance status, and comorbidity provide a crude discrimination of prognosis in most tumor types, including nonsmall-cell lung cancer (NSCLC). These variables are surrogate markers of clinical behavior and could be useful for predicting patient prognosis and selecting appropriate anticancer treatments [Citation1]. For instance, mediastinal lymph node involvement and the number of metastatic lymph nodes are important negative prognostic factor in surgically resected stage IIIA NSCLC [Citation2]. Additionally, visceral pleural invasion was observed more frequently in biologically aggressive NSCLCs, and by multivariate analysis, this invasion proved to be a significant independent predictor of grim prognosis, with or without lymph node involvement [Citation3]. Therefore, in most NSCLC cases, the therapeutic strategy is based on the tumor stage and on the performance status of the patient at diagnosis.
However, recent data suggested that the histologic subtype influences the efficacy or toxicity of anticancer treatments. This differential therapeutic efficacy is well documented for pemetrexed in advanced or metastatic NSCLC: a phase III trial showed that patients with nonsquamous histology had a survival benefit when treated with cisplatin/pemetrexed versus cisplatin/gemcitabine, while the reverse was observed in patients with squamous histology [Citation4]. On the basis of these results, the Food and Drug Administration (FDA) and European Medicine Agency (EMEA) approved pemetrexed for the use in the first-line treatment of advanced nonsquamous NSCLC.
Most recently, the treatment of NSCLC has been revolutionized by the introduction of novel drugs designed to target specific molecular factors implicated in cancer cell proliferation. These novel targeted therapies are based on advances in our understanding of key cellular networks and nodal points involved in tumor development and progression [Citation5]. Genomic studies have indeed highlighted the importance of ‘driver’ somatic alterations that activate crucial oncoproteins originating cancers with an addictive dependency. Therapeutic agents specifically designed to intercept these deregulated oncoproteins have demonstrated to be effective treatments in such ‘oncogene-addicted’ tumors. In particular, the epidermal growth factor receptor (EGFR) has been successfully targeted either by monoclonal antibodies or small molecules inhibiting the tyrosine kinase (TK) domain. The monoclonal antibody cetuximab blocks the extracellular domain of EGFR, thereby competing with the ligands, and has been approved as first-line treatment combined with platinum-based chemotherapy in EGFR-positive NSCLC patients with good performance status [Citation6]. The EGFR tyrosine kinase inhibitor (TKI) gefitinib has been approved by FDA and EMEA as upfront therapy replacing chemotherapy in late-stage NSCLC patients harboring activating EGFR mutations [Citation7]. The manageable toxicity, along with its efficacy, make erlotinib an important option also as maintenance therapy, and erlotinib and gefitinib are the only drugs of proven efficacy in the third-line setting for NSCLC [Citation8]. Another example of targeted therapy is the anaplastic lymphoma kinase (ALK) inhibitor crizotinib, which has been approved by FDA for the treatment of locally advanced or metastatic NSCLCs with the fusion-type protein kinase echinoderm microtubule-associated protein-like 4 (EML4)–ALK translocation [Citation9]. This traslocation occurs in up to 5% of NSCLC tumors as a result of a small inversion within the short arm of human chromosome 2.1. Fusion of EML4 with ALK enables ALK to dimerize ligand independently, resulting in an aberrantly active protein. Aberrant expression of the EML4-ALK fusion protein activates several pathways important for cell survival and cell proliferation such as PI3K, JAK3/STAT, and Ras/Mek/Erk pathways [Citation10]. The fast-tracked FDA approval of crizotinib, as well as of the second-generation ALK inhibitor ceritinib, which inhibits two of the most common ALK mutants that confer resistance to crizotinib, L1196M and G1202R,[Citation11] is due to the excellent clinical trial design and should set an example for future cancer drug development. The efficacy of these compounds in NSCLC patients is indeed based on well-researched mechanisms and proven to be a solid choice as therapy for NSCLC.
However, the clinical success of the new biologically targeted agents has been limited by early intrinsic resistance (also known as innate or de novo resistance) or fast adaptive tumor responses (acquired resistance) to targeted anticancer therapies, because of their inability to fully block the cancer-relevant signaling pathways [Citation12]. Furthermore, despite the high selectivity of TKIs, in daily clinical practice, several previously unknown and sometimes unpredictable adverse events are emerging. These effects are typically related with targets which are also present in normal tissues, such as the skin and mucous membranes, the gastrointestinal tract, the cardiovascular, respiratory, and neurologic systems [Citation13]. Clinicians must be challenged to recognize and manage these ‘novel’ adverse events because, even if in the majority of cases they are of mild–moderate grade and reversible on treatment cessation, they could deeply influence patients’ quality of life. The main problem of current cancer therapies is that many patients are indeed receiving a toxic and often expensive treatment without any benefit.
Pharmacogenetics can be defined as the study of how the actions of and reactions to drugs vary because of genetic factors regulating individual and tumoral drug pharmacokinetics and pharmacodynamics. Therefore, pharmacogenetics of both conventional chemotherapeutic and targeted agents should provide key insights on the intersubject treatment-response and toxicity variability. Inherited genetic polymorphisms, including single-nucleotide polymorphisms (SNPs) that affect either expression or functionality of important enzymes and/or targets in the metabolism and activity of anticancer drugs have been extensively investigated as prognostic/predictive factors in different tumor types, including NSCLC [Citation14]. The large majority of these studies applied a candidate gene approach based on ‘a priori’ knowledge of gene function, leading to often conflicting results that are difficult to replicate [Citation15]. The present review summarizes the main findings on NSCLC pharmacogenetics, critically reappraising the most important factors affecting the data on polymorphisms correlated with drug activity or toxicity. Because of their current use in the treatment of NSCLC, we especially report the clinical trials and pharmacogenetic determinants of a classical chemotherapeutic compound, pemetrexed, and of the widely used EGFR-TKIs gefitinib and erlotinib. Other issues include the development of new technologies to decipher pharmacogenetic markers of chemosensitivity and/or resistance that might be applied to the patient before and during treatment using samples from surrogate tissues. Furthermore, we highlight the most recent and clinically relevant aspects of genetic analyses of NSCLC from the perspective of the application in the clinical practice, reporting which specimens and methods should be used to conduct appropriate pharmacogenetic studies, as described in the following chapters.
2. Pharmacogenetics of NSCLC: which patients and specimens?
Assessing germline genetic polymorphisms as either predictive or prognostic markers is very appealing, especially in the NSCLC advanced cancer setting, when diagnosis is usually done from small needle biopsy samples, and tumors are either not resected or resected after chemotherapy, so that the handling of patient material can be problematic [Citation16]. Polymorphisms are indeed inherited genetic variants harbored by all the cells, and the biological material required for pharmacogenetic studies is therefore germline DNA. This DNA can be easily extracted from blood samples as well as from other normal cells of the body, such as cells from internal cheek mucosa, which can be obtained with a noninvasive procedure through a smooth brush [Citation17]. Thus, pharmacogenetic analyses are easier to adopt in the routine clinical setting than tumor gene expression studies, which need core needle biopsies of patient’s tumors with laser microdissection and subsequent sophisticated infrastructure. However, some pharmacogenetic studies evaluated tumor material, obtained from biopsy or surgery, which was stored as fresh frozen samples or archived in pathology laboratories after formalin fixation and embedding in paraffin blocks [Citation18]. The tumor cores in these samples are almost always surrounded by nontumor areas that should be used for DNA extraction. Despite numerous important genomic alterations that characterize tumor DNA, the ‘ordinary’ polymorphisms still exist in the tumor genome. However, the risk of loss of heterozygosity may introduce a critical bias and alter the conclusions of a study dedicated, for instance, at the identification of the polymorphisms of a gene expressed in the liver and involved in drug activation or detoxification, such as cytidine deaminase for gemcitabine [Citation19]. Conversely, it might be interesting to know the tumor as well as the germinal genotype of polymorphisms of DNA repair genes, because the DNA repair involved in resistance to alkylating drugs actually occurs within the cancer cells [Citation20].
Of note, recent reports have shown that lung cancers can be characterized by substantial heterogeneity [Citation21]. A recent study performing multiregion whole-exome sequencing on 48 tumor regions from 11 localized resected lung adenocarcinomas identified 76% of all mutations and 20 of 21 known cancer gene mutations in all regions of individual tumors, suggesting these represent early events in tumorigenesis. Parallel deep sequencing was able to detect gene mutations in all the regions and characterize heterogeneity more accurately [Citation22]. These results support the analysis of normal cells for pharmacogenetic studies, as well as further studies on circulating tumor DNA [Citation23,Citation24], which might reflect better the entire tumor genome and can offer a more feasible means of comparing tissue analysis and evaluating tumor heterogeneity at diagnosis or during treatment in order to better tailor therapeutic strategies with targeted agents or drug combinations.
3. Pharmacogenetics of NSCLC: which genes, copy number variations/alterations and polymorphisms?
The unsatisfactory results of currently available therapies for NSCLC might reflect the lack of knowledge on the way by which molecular abnormalities affect tumor responsiveness to anticancer agents. A few studies evaluated copy number variations/alterations correlated to NSCLC outcome. In particular, copy number gains on chromosome 8q21-q24.3 were statistically significantly more frequent in long-term than in short-term survivors affected by lung adenocarcinoma, whereas in squamous cell carcinoma, gains on chromosome 14q23.1–24.3 were associated with shorter overall survival (OS), and losses in 14q31.1–32.33 correlated with longer OS [Citation25]. Since histological subtypes are associated with differential outcome and therapeutic response [Citation4], we might speculate that these genetic aberrations could affect drug activity. Importantly, cigarette smoking, which is one of the main causes of NSCLC and has also been related to prognosis in NSCLC patients treated with cisplatin and gemcitabine or cisplatin and pemetrexed [Citation26], leads to more copy number alterations, which may be mediated by the genome instability [Citation27]. A recent study showed that nicotinic acetylcholine receptor alpha3, alpha5, and beta4 genes (CHRNA3, CHRNA5, and CHRNB4) cluster at the 15q25.1 lung cancer susceptibility locus, and four variants (rs3829787, rs3841324, rs588765, and rs3743073) were associated with differential levels of genetic aberrations, while three haplotypes were associated with CHRNA5 mRNA levels, susceptibility to NSCLC, smoking-related DNA alterations, and TP53 mutations [Citation28]. Genetic differences resulting in interindividual variation in DNA repair capacity may indeed account for susceptibility of the lung cells to the genotoxic agents leading to somatic mutations, and individuals who smoke and have polymorphisms that may result in deficient DNA repair are at a greater risk of TP53 mutations. Furthermore, the interaction between genetic variations such as SNPs and copy number variations and somatic mutations in some of the tumor most important genes might affect response to specific anticancer drugs, and several studies correlated cisplatin and DNA damage and repair with TP53 mutations and p53 activity [Citation29]. Many efforts have been therefore invested to identify pharmacogenetic biomarkers that can be used to predict sensitivity or resistance to cisplatin, including TP53 and DNA repair pathway genes [Citation20]. However, the present review focuses on examples on how genetics might affect drug response of the conventional chemotherapeutic agent pemetrexed as well as of the anti-EGFR targeted agents gefitinib and erlotinib, as described in the following paragraphs.
3.1. Pharmacogenetics of pemetrexed
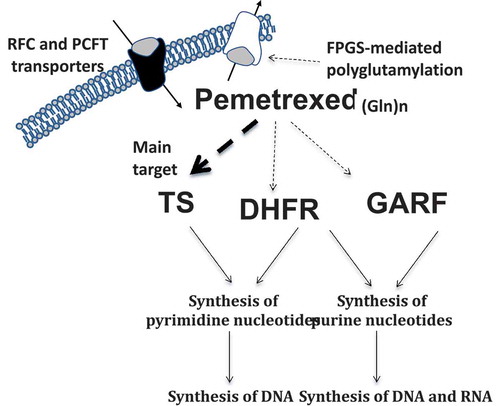
Pemetrexed enters the cell via the reduced folate carrier (RFC) or proton-coupled folate transporter and is converted by folypolyglutamate syntethase to a series of active polyglutamate derivatives inhibiting several folate-dependent enzymes such as thymidylate synthase (TS), dihydrofolate reductase (DHFR), glycinamide ribonucleotide formyl transferase (GARFT), and to a lesser extent, aminoimidazole carboxamide ribonucleotide tranformylase and CI-tetrahydrofolate synthase (). Its activity leads to fully reduced folates depletion, thus resulting in disruption of purines and pyrimidines synthesis [Citation30]. The polyglutamylation leads to extended intracellular retention of the compound, resulting in more prolonged efficacy. While pemetrexed-mediated inhibition of GARFT is weak, TS inhibition interrupts the tetrahydrofolate oxidation with consequential lack of DHFR activity [Citation31]. Preclinical data showed a significant correlation between TS mRNA and protein upregulation with reduced sensitivity to pemetrexed in NSCLC cell lines [Citation32,Citation33]. Sensitivity to pemetrexed was also associated with mRNA levels of genes involved in the mechanism of action of this drug in freshly explanted human tumor specimens. In particular, low TS mRNA, as well as reduced levels of GARFT and DHFR mRNA, significantly correlated with chemosensitivity to pemetrexed [Citation34]. Several other studies showed that the expression of TS protein was correlated with outcome in NSCLC specimens from patients treated with pemetrexed-based chemotherapy, particularly in nonsquamous carcinomas [Citation35,Citation36]. The different clinical results obtained with pemetrexed in various histological subtypes of NSCLC [Citation4] might indeed be explained with the differential TS expression, which is higher in squamous cell carcinomas than in adenocarcinomas [Citation37]. These data have been corroborated by a recent meta-analysis in a total of 526 patients from the eight studies, showing that objective response rate for pemetrexed-containing chemotherapy was significantly higher in patients with TS low expression. However, although patients with low expression of TS have a longer median OS, this difference was not statistically significant [Citation38]. These conflicting results might be due to relatively small/heterogeneous sample size, different methodologies, treatment heterogeneity, and retrospective nature of most studies.
Figure 1. Key factors in the transport, activation, and activity of pemetrexed. Dashed lines denote inhibitory effect of drug/metabolites on cellular enzymes/transporters. Abbreviations. DHFR: Dihydrofolate reductase; FPGS: Folylpolyglutamate synthetase; GARFT: Glycinamide ribonucleotide formyl transferase; Gln: Polyglutamate; PCFT: Proton-coupled folate transporter; RFC: Reduced folate carrier; TS: thymidylate synthase.
More recently, Sun and collaborators [Citation39] reported data from an observational phase II study where they prospectively investigated the correlation between tumor baseline expression of TS and outcome in patients with nonsquamous NSCLC randomly assigned to either pemetrexed/cisplatin or gemcitabine/cisplatin treatment. Low-TS protein levels were predictive of significantly higher response rates and longer progression-free survival (PFS) in patients treated with pemetrexed/cisplatin compared to the gemcitabine/cisplatin group. However, regarding the same treatment allocation, high TS expression was not correlated to the outcome. A power analysis for the number of enrolled patients as well as a consensus regarding the scoring values to assess protein expression are urgently needed to improve experimental reproducibility and correct interpretation of results. Similarly, the optimization and standardization of technical procedures are crucial to validate the best biomarkers for personalized chemotherapy.
A phase III multicenter, randomized study ITACA (International TAilored Chemotherapy Adjuvant) has been started to prospectively validate the role of TS in pemetrexed-based chemotherapy response [Citation40]. In this trial, expression of TS and ERCC1 is assessed by PCR on tumor specimens before randomization in all the enrolled patients. Within 45 days post-surgery, patients in each genetic profile are randomized to receive either a standard chemotherapy selected by the investigator (cisplatin/vinorelbine, cisplatin/docetaxel, or cisplatin/gemcitabine) or the following experimental treatments: (1) high ERCC1/high TS, single agent paclitaxel; (2) high ERCC1/low TS, single agent pemetrexed; (3) low ERCC1/high TS, cisplatin/gemcitabine; (4) low ERCC1/low TS, cisplatin/pemetrexed. All regimens are administered for a total of 4 cycles on a 3-weekly basis, and OS has been indicated as primary endpoint. However, further prospective randomized trials, with a control and a genotypic arm, should be performed in patients affected by advanced/metastatic NSCLC.
Remarkably, TS expression is regulated by multiple mechanisms occurring at transcriptional, post-transcriptional, and translational levels [Citation41]. These physiological mechanisms are associated with cell proliferation and apoptosis, which are often altered in tumors; therefore, we believe that functional studies to evaluate key factors modulating TS status are necessary before larger pharmacogenetics prospective trials are launched. TS may indeed have a mixed role as prognostic and predictive biomarker, possibly depending on drug and disease [Citation30].
Functional polymorphisms such as polymorphic tandem repeats located in the TS enhancer region (TSER) have also been investigated. Previous studies reported a 2.6-fold higher TS expression with 3 copies (TSER*3) of the R than with 2 copies (TSER*2) [Citation42]. The variable number of repeats was associated with outcome after pemetrexed treatment in patients with different malignancies, including lung cancers [Citation43,Citation44]. However, these data were not always confirmed in other studies in NSCLC [Citation45–Citation47]. In particular, the randomized phase II trials NVALT-7 and GOIRC-02.2006, which included the largest retrospective pharmacogenetic study performed in NSCLC patients treated with pemetrexed, did not confirm the correlation of TSER with outcome [Citation47]. Conversely, the homozygous variant methylenetetrahydrofolate reductase T677 T was reported to correlate with a significantly longer PFS compared to patients with the wild-type or heterozygous genotype in the same study. Since methylenetetrahydrofolate reductase is an important regulator of the folate homeostasis, the polymorphic variant was suggested to favor an increased inhibition of TS by pemetrexed. However, this result was not observed in 45 adenocarcinoma patients treated with pemetrexed [Citation44].
Pemetrexed activity might indeed be affected by other mechanisms of antifolate resistance such as loss of RFC or proton-coupled folate transporter, causing impaired uptake: increased drug efflux due to overexpression of ATP-driven multidrug resistance proteins, decreased folypolyglutamate syntethase expression and/or inactivating mutations or alternative splicing defecting antifolate polyglutamylation, and increased expression of γ-glutamyl hydrolase (GGH) [Citation30,Citation44]. However, none of these factors has yet been validated in NSCLC [Citation15]. Recent pharmacogenetic studies in NSCLC suggested the potential predictive role of SNPs in RFC and GGH genes. A study evaluating the efficacy of combining pemetrexed and bevacizumab in 48 patients suggested the correlation of RFC-exon6-SNP and PFS. Additionally, a trend toward an association with PFS was associated with the GGH IVS7(1478) SNP, whereas the GGH IVS2(1307)CC genotype was associated with significantly longer OS [Citation48]. In contrast, pharmacogenetic analyses in a randomized phase II trial aiming to optimize the administration schedule of pemetrexed and gemcitabine in 54 chemotherapy-naive patients with advanced NSCLC did not show any correlations with outcome for GGH IVS7(1478) and IVS2(1307) [Citation49]. However, the latter trial supported the association of RFC-exon6-SNP with outcome following pemetrexed treatment [Citation49], which was also reported in the recent research by Li and collaborators [Citation44].
Future pharmacogenetic studies should be integrated with the analysis of active drug metabolites, such as intracellular pemetrexed polyglutamates, which might lead to a more complete picture on the pharmacokinetics and pharmacodynamics of pemetrexed and enable better prediction of individual treatment response.
3.2. Pharmacogenetics of EGFR-TKIs
Several germline DNA variations of EGFR and other genes have been associated with clinical outcome, and this section focuses on the relationship between these candidate germline polymorphisms () and the response and toxicity to the EGFR-TKIs [Citation50]. However, studies on polymorphisms affecting their outcome have the potential to be extended to cover TKIs of similar structure and activity, including second- and third-generation EGFR inhibitors with increasing clinical application, such as afatinib, rociletinib, and AZD9291.[Citation51,Citation52]
Table 1. Key genetic polymorphism associated with differential outcome and toxicity in NSCLC patients treated with gefitinib and/or erlotinib.
Most pharmacogenetic studies evaluated SNPs in the region regulating the expression of the EGFR gene, which are located within the 5ʹ-flanking region and intron-1. Both the EGFR −191C/A and −216G/T SNPs are located in the transcriptional start site of the promoter region, including multiple nuclear regulatory affinity sites. The −191C/A polymorphism has been correlated with enhanced EGFR expression and activity, while the A-G variant, leading to the substitution of an arginine with a lysine at codon 497 (R497K), has been associated with reduction of EGFR activity [Citation53,Citation54]. The −216G/T genotype is at the binding site for the transcription factor Sp1, and the T allele increased EGFR mRNA expression [Citation55]. The −216G/T, −191C/A, and R497K EGFR polymorphisms were evaluated in a study conducted in advanced NSCLC patients treated with gefitinib, showing a significant association of the −216G/T variant with longer PFS. This variant was also associated with significantly higher rates of stable disease/partial response, and a significantly higher risk of treatment-related rash and diarrhea [Citation56]. However, more recent studies in Japanese cohorts treated with gefitinib as first-line or adjuvant therapy did not show significant associations [Citation57,Citation58], while lower response rates to gefitinib were observed in Caucasian patients harboring the G/C haplotype [Citation59].
Another EGFR region with relevant polymorphisms is located in the intron-1, characterized by 14 to 21 CA-repeats; with shorter alleles of a CA-dinucleotide repeat polymorphism in intron-1 associated with enhanced EGFR transcription, improved response rates, and longer OS after gefitinib treatment in different ethnicity cohorts [Citation56,Citation58–Citation68]. Most studies reported a better response to gefitinib, both in Japanese and Caucasian patients. For instance, in 70 NSCLC Chinese patients treated with gefitinib, a significantly higher response rate was associated with shorter CA-repeat status (defined as any allele less than 16). These patients had also higher EGFR expression and prolonged OS compared to those with long CA [Citation67]. Similarly, shorter CA repeats status (16 or less) was associated with significantly longer PFS and OS in another study in 92 Caucasians NSCLC patients treated with gefitinib [Citation56]. However, in the largest pharmacogenetic analysis in NSCLC Caucasian patients treated with gefitinib (n = 175), no association of the EGFR intron-1 CA-repeat status with clinical outcome was observed [Citation59]. Furthermore, the prospective analysis of the prognostic and predictive value in the SATURN study in 889 advanced NSCLC patients did not show a significant association between CA-repeat polymorphisms, EGFR expression and gene copy number, and PFS after erlotinib treatment [Citation69].
Other polymorphisms that have been correlated to EGFR-TKIs outcome include variations in the EGFR downstream signaling pathways such as AKT1, as well as the DNA repair genes and those of the genes encoding for the drug transporter ABCG2, which has been shown to be active in removing gefitinib from the cell [Citation70]. The haplotype including two functional polymorphisms (AKT1-SNP3 and SNP4) was associated with lower Akt protein levels in tissues from Caucasians and with the lowest apoptotic response of EBV-transformed lymphoblastoids to radiation [Citation71,Citation72]. In 96 Caucasian patients treated with gefitinib, the AKT1-SNP4 A/A genotype was correlated with shorter OS [Citation73], while AKT1-rs2498804 GT and GG alleles were associated with the occurrence of brain metastases [Citation74]. Similarly, a recent study in Korean NSCLC patients showed that other AKT1 polymorphisms could be used as prognostic markers for patients with early-stage NSCLC, suggesting that several genetic variations in the PI3K/AKT pathway may be prognostic and/or predictive factors of response to different drugs [Citation75]. However, these results have still to be validated in a larger cohort of patients, in prospective multicenter trials, as well as additional case–control studies.
At clinically achievable concentrations (≤1 µM), gefitinib is an ABCG2 substrate, and ABCG2 is highly expressed in the gastrointestinal tract where it participates in the uptake of several xenobiotics, including oral EGFR-TKIs [Citation76]. This member of the ATP-binding cassette (ABC) transporter family could play a key role in the absorption and elimination of gefitinib, which might therefore be affected by several common SNPs in the ABCG2 gene that might affect ABCG2 protein expression, function, and localization [Citation77]. In particular, the ABCG2 421C/A polymorphism resulting in a glutamine to lysine amino acid change at position 141 (Q141K) has been correlated with the reduction of ABCG2 protein expression and/or activity and with increase accumulation of both gefitinib and erlotinib [Citation78]. However, no correlation between ABCG2 421C/A polymorphism and protein expression, as well as with outcome after gefitinib treatment, was observed in 50 NSCLC tissues [Citation79]. Similarly, the SATURN trial, evaluating erlotinib in 242 advanced NSCLC patients, did not report significant correlation between response to erlotinib treatment and the ABCG2 421C/A SNPs, whereas the EGFR 216T/T genotype was related with the development of skin toxicity [Citation53].
Several other studies evaluated the correlation between selected polymorphisms and toxicity induced by EGFR-TKIs. Although targeted agents are generally less toxic than traditional anti-neoplastic agents, EGFR-TKIs are indeed associated with a number of bothersome adverse effects, such as rash and diarrhea. At present, little is known about the etiology of these adverse effects, but the high interpatient variability might be explained by the pharmacogenetic heterogeneity of patients [Citation80]. In a study in 52 NSCLC patients treated with gefitinib the intron-1, CA repeat variant was correlated with grade 2–3 skin rash, observed in 21% of patients with 19–22 repeats, 31% of patients with 15–18 repeats, and 71% of patients with less than 15 repeats [Citation81]. Other studies reported the association of the EGFR 191C/A and A/A, the EGFR 216G/G, and the R497K A/A variants with a significantly higher risk of diarrhea in NSCLC patients treated with gefitinib [Citation56]. These data support a role of high EGFR expression in the intestinal lumen associated with the EGFR promoter polymorphism variants among the causes of diarrhea, as also reported by Rudin and collaborators [Citation82]. However, diarrhea was also correlated with the ABCG2 421C/A variant: 44% of gefitinib-treated NSCLC patients heterozygous for ABCG2 421C/A presented this adverse effect [Citation83]. Conversely, no correlation between gefitinib-induced toxicity and ABCG2 421C/A was found in a population of 94 Caucasian NSCLC patients, where moderate–severe diarrhea correlated with the ABCG2 15622C/T polymorphism and the ABCG2 (1143C/T, −15622C/T) haplotype [Citation79].
Finally, both gefitinib and erlotinib are metabolized by the CYP3A4, CYP3A5, and CYP1A isozymes, while CYP1A2 is involved in the metabolism of erlotinib, but not of gefitinib [Citation84]. Since all these CYPs are polymorphic, the distribution of specific variant CYP alleles might explain the different pharmacokinetics of TKIs in the patients. CYP3A4 polymorphisms have been indeed associated with skin rash in erlotinib-treated patients: individuals with lower CYP3A4 expression (because of the A/A variant) were more likely to develop rash than those with higher CYP3A4 levels (A/G and G/G variants) [Citation82]. In the same study, the CYP3A5*3 G polymorphism was also associated with grade ≥2 rash and diarrhea. However, all these associations were marginal, and further studies on the role of CYP3A4 and CYP3A5 polymorphisms in determining activity and toxicity of EGFR-TKIs are warranted.
In conclusion, despite the intriguing findings of several studies, the small sample size together with the interethnic differences, and the retrospective nature of most studies, make it difficult to draw any clear conclusions regarding the role of several pharmacogenetic biomarkers in determining the clinical outcome or toxicity in gefitinib and erlotinib treatment. Hopefully, the more accurate planning of randomized prospective trials, the increased insights of molecular mechanisms underlying drug distribution/activity, and the use of novel technologies, including whole genome approaches, may provide fundamental tools to transfer the knowledge generated though pharmacogenetic studies to clinical practice.
4. Pharmacogenetics of NSCLC: which methods and trials?
As exemplified in the previous paragraphs, controversial results raise some reluctance on the potential use of pharmacogenetic biomarkers in patient’s management. This could be due to several issues about clinical and laboratory methodologies.[Citation83,Citation85]
A frequent question concerns the way of considering the heterozygotes relatively to wild-type and variant homozygotes [Citation17]. When the size of the population is large enough and when the functional effect of the polymorphism is not known, it is certainly better to analyze independently the three genotypes, with the assumption that each variant allele contributes to the phenotype. However, when it is known that the presence of one copy of the variant allele is sufficient to determine the phenotypic character (and has therefore a dominant role), one can regroup heterozygotes with variant homozygotes; while when it is necessary that the two variant alleles are expressed to determine the phenotype (which is consequently recessive), the heterozygotes should be regrouped with the common homozygotes. Unfortunately, not all the studies enrolled an adequate number of patients and not all the studied polymorphisms are functional, nonsynonymous polymorphisms, which result in a difference in the expression or activity of the encoded protein.
Whereas pharmacokinetic evaluation of any drug is required during clinical trials, relationships between gene polymorphisms and pharmacological activity are often discovered fortuitously after the drug has been prescribed for many years. Hopefully, in the future, more systematic pharmacogenetics studies should be performed during the phase II and III trials of clinical development, which include several hundreds of patients. Pharmacogenetic studies must indeed be large in order to have enough statistical power to detect a gene effect, a treatment effect and a drug–gene interaction [Citation86]. The power to detect a statistical interaction depends on the number of SNPs, the allelic frequencies, and the type of study design. The analysis of multiple polymorphisms require a correction for multiple testing, such as the Bonferroni correction, in which the P value that is used for one test is divided by the total number of tests in the analysis [Citation87]. However, the use of the Bonferroni correction may be considered too conservative because many polymorphisms are in linkage disequilibrium, which may mask their effects and increase type II errors. Furthermore, since many of the pharmacogenetic trials included a small number of subjects and are underpowered to detect a drug–gene interaction, the Bonferroni correction may null the study results [Citation88].
In most pharmacogenetic studies, candidate polymorphisms are analyzed because of the chemical and pharmacological characteristics of the drug, considering the molecules involved in the metabolism or the molecular target. However, in vitro models may orientate the choice of the polymorphisms of interest. In particular, the panel established by the National Cancer Institute, including 60 human tumor cell lines whose chemosensitivity to thousands of compounds, has been evaluated by several groups [Citation89]. More recently, two alternative panels of cell lines have been published, the Cell Line Encyclopedia and the Sanger collection [Citation90,Citation91], including 947 and 639 cell lines, respectively; but less compounds have been tested in these models. In addition, the new molecular biology tools allow the creation of isogenic cell lines, differing only by the presence of the variation studied in the gene of interest. Such isogenic cell lines may be subjected to phenotypic characterization in vitro and in vivo when they can be xenografted in immunodeficient mice.
However, the availability of genome-wide approaches enables original discoveries of novel and less obvious genes to be detected, especially in the area of drug-target genetics, which is more complex and less well understood than the pharmacogenetics of drug metabolism [Citation92]. These novel techniques include genome-wide association studies (GWAS) and massively parallel or next generation sequencing (NGS). GWAS investigate the entire genome and are not based on preliminary information about the involvement of particular gene or chromosomal locus in drug effects. However, an important limitation of this analysis is that it allows the identification of SNPs and other DNA variants which are associated to drug activity, but cannot specify which genes are casual, with the risk of a high rate of false positive data. A high number of samples is therefore essential to obtain a sufficient statistical power. For instance, a GWA of 307.260 SNPs has been performed in 327 advanced NSCLC patients treated with platinum-based therapy [Citation93]. This study identified the strong association of the rs1878022 SNP, located within an intron of the chemokine-like receptor 1 gene, with shorted survival. Importantly, these data were validated in two independent cohorts of 315 and 420 patients. More recently, the rapidly decreasing prices of complete genome sequencing with NGS have provided a realistic alternative to genotyping array-based GWA studies [Citation94]. Although GWAS and NGS have the potential to dramatically accelerate biomedical research also in the pharmacogenetics field, these technologies are still limited by the lack of standardized protocols and bioinformatics infrastructures that can integrate different information and extract clinically useful data. A standardization of these methodologies will be indeed critical to perform analysis on large cohorts of patients and/or sharing data through common websites in order to create ‘virtual multicentric trials’.
5. Expert opinion
Advances into the knowledge of the genetics of lung cancer have recently identified key biological processes and molecular targets for antitumor treatment. Novel agents targeting these aberrant processes have revolutionized the management of specific molecular subsets of NSCLC and have greatly contributed to recent improvements in survival rates. For example, EGFR mutant and EML4-ALK fusion status, which are detected in approximately 15% and 4% of lung adenocarcinomas, predict response to therapies with selective inhibitors. These results led to the approval of EGFR-TKIs and the ALK inhibitors as first-line treatments in molecularly selected NSCLC patients [Citation8,Citation9].
However, the oncologists are still facing relevant interindividual variability in drug activity as well as the occurrence of relevant drug adverse events. Therefore, to improve the rational use of this emerging arsenal of highly selective, targeted cancer therapeutics, as well as of the conventional chemotherapeutic drugs, the histopathological assessment of tumors should be associated with a refined pharmacologenetics evaluation.
The adoption of pharmacogenetics in the clinical practice is indeed a high research priority in oncology. Although the list of pharmacogenomic biomarkers in drug labeling include several drugs used for NSCLC (), and a number of important genetic determinants have been identified thus far in different tumor types, including NSCLC, the clinical relevance of most germ-line gene variants currently remains unconfirmed, and such data have not yet been translated into a better therapeutic management [Citation83].
Table 2. Anticancer drugs used in the clinical setting for the treatment of NSCLC that have pharmacogenomic information in their labeling.
The controversial results about the correlation of specific pharmacogenetic markers with outcome could be due to a number of reasons, such as (1) the small number of (heterogeneous) patients; (2) the lack of appropriately designed prospective studies; (3) the lack of standardized analytical methodologies and quality controls; and (4) the intra-tumor heterogeneity as well as other complex factors, including reversible, epigenetic factors, such as mRNA, microRNA, and factors affecting DNA methylation [Citation96,Citation97], which might foster tumor adaptation and therapeutic failure. For instance, a recent meta-analysis-based approach evaluating 12 cohorts of 1069 tumor-node-metastasis stage I lung adenocarcinoma patients showed that a 4-gene classifier, using the expression levels of BRCA1, HIF1A, DLC1, and XPO1, provided independent prognostic stratification of stage IA and stage IB patients beyond conventional clinical factors [Citation98].
Most of the limitations of the previous pharmacogenetic studies should be overcome by the well-designed prospective clinical trials in which a direct comparison is performed between patient treatments selected on the basis of standard criteria vs. treatment selection suggested by patient/tumor genetic characteristics. Moreover, to evaluate the heterogeneity and possible evolution of cancer cells, these new clinical protocols should include multiple and repeated biopsies of the single tumor or novel liquid biopsies [Citation99].
The lack of correlation of candidates emerging from pharmacogenetic studies with the main pharmacokinetics/pharmacodynamics parameters is another obstacle to the successful clinical application of novel biomarkers. However, the recent discovery of microRNA may improve the knowledge about the complex control of gene expression, and help to clarify the role of different signaling pathways, involved in oncogenesis and drug resistance, in different tumor types, including NSCLC [Citation100].
Finally, widespread use of novel technologies, including genome-wide approaches, such as GWAS and NGS, has the potential to deepen patient’s genetic information in order to predict optimal dosage for a drug with a narrow therapeutic index, as well as to select the most appropriate pharmacological agent for a given patient. All these innovations represent therefore essential tools to improve pharmacogenetic studies, which will hopefully lead to optimization of novel and currently available treatments in selected NSCLC patients.
Article Highlights
Despite the discovery of all the successful targeted therapies directed against specific alterations of oncogenic pathways, nonsmall-cell lung cancer (NSCLC) is still the leading cause of cancer-related death, with overall 5-year survival rate of about 15%.
Pharmacogenetics intends to identify relationships between gene polymorphisms and drug activity (in terms of both efficacy and toxicity), with the aim of proposing a rational individual drug prescription.
Examples of pharmacogenetic determinants are the polymorphic tandem repeats located in a drug target, such as the TS enhancer region (TSER), which were associated with outcome after pemetrexed treatment and the ABCG2 421C/A singlenucleotide polymorphisms which has been correlated with the reduction of ABCG2 protein expression and/or activity and with increased accumulation of gefitinib and erlotinib.
Up to now, most pharmacogenetic studies led to conflicting results that are difficult to translate in the clinical setting.
Future pharmacogenetics studies should improve both (1) laboratories methodologies, including standardized techniques of sample collection/processing and mathematically robust methods to analyze data, and (2) clinical methodologies, including large prospective randomized clinical trials, in homogeneous population, with appropriate design of customized therapy and powered statistical analysis.
New technologies will foster pharmacogenetic by moving from candidate gene methods, based on a priori knowledge of gene function, toward a genome-wide approach.
Declaration of Interest
This work was supported by Associazione Italiana per la Ricerca sul Cancro (Start-Up grant and iCARE grant, to E Giovannetti and LG Leticia), and Cancer Center Amsterdam (2012 and 2015, to E Giovannetti and GJ Peters). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- Dietel M, Jöhrens K, Laffert M, et al. update on predictive molecular pathology and its role in targeted cancer therapy: a review focussing on clinical relevance. Cancer Gene Ther. 2015;22:417–430.
- Andre F, Grunenwald D, Pignon JP, et al. Survival of patients with resected N2 non-small-cell lung cancer: evidence for a subclassification and implications. J Clin Oncol. 2000;18:2981–2989.
- Shimizu K, Yoshida J, Nagai K, et al. Visceral pleural invasion is an invasive and aggressive indicator of non-small cell lung cancer. J Thorac Cardiovasc Surg. 2005;130:160–165.
- Scagliotti GV, Parikh P, Von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543–3551.
- Gross S, Rahal R, Stransky N, et al. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–1789.
- Pirker R, Pereira JR, Szczesna A, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373:1525–1531.
- Sebastian M, Schmittel A, Reck M. First-line treatment of EGFR-mutated nonsmall cell lung cancer: critical review on study methodology. Eur Respir Rev. 2014;23:92–105.
- Reck M, Mok T, Wolf J, et al. Reviewing the safety of erlotinib in non-small cell lung cancer. Expert Opin Drug Saf. 2011;10:147–157.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703.
- Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–4283.
- Muller IB, De Langen AJ, Honeywell RJ, et al. Overcoming crizotinib resistance in ALK-rearranged NSCLC with the second-generation ALK-inhibitor ceritinib. Expert Rev Anticancer Ther. 2015 Jan 4:1–11. [Epub ahead of print]
- Rolfo C, Passiglia F, Castiglia M, et al. ALK and crizotinib: after the honeymoon…what else? Resistance mechanisms and new therapies to overcome it. Transl Lung Cancer Res. 2014;3:250–261.
- Novello S, Capelletto E, Cortinovis D, et al. Italian multicenter survey to evaluate the opinion of patients and their reference clinicians on the “tolerance” to targeted therapies already available for non-small cell lung cancer treatment in daily clinical practice. Transl Lung Cancer Res. 2014;3:173–180.
- Hildebrandt MA, Gu J, Wu X. Pharmacogenomics of platinum-based chemotherapy in NSCLC. Expert Opin Drug Metab Toxicol. 2009;5:745–755.
- Giovannetti E, Toffalorio F, De Pas T, et al. Pharmacogenetics of conventional chemotherapy in non-small-cell lung cancer: a changing landscape? Pharmacogenomics. 2012;13:1073–1086.
- Igbokwe A, Lopez-Terrada DH. Molecular testing of solid tumors. Arch Pathol Lab Med. 2011;135:67–82.
- Robert J, Le Morvan V, Giovannetti E, et al. On the use of pharmacogenetics in cancer treatment and clinical trials. Eur J Cancer. 2014;50:2532–2543.
- Wright CM, Larsen JE, Colosimo ML, et al. Genetic association study of CYP1A1 polymorphisms identifies risk haplotypes in non small cell lung cancer. Eur Respir J. 2010;35:152–159.
- Tibaldi C, Giovannetti E, Tiseo M, et al. Correlation of cytidine deaminase polymorphisms and activity with clinical outcome in gemcitabine-/platinum-treated advanced non-small-cell lung cancer patients. Ann Oncol. 2012;23:670–677.
- Rose MC, Kostyanovskaya E, Huang RS. Pharmacogenomics of cisplatin sensitivity in non-small cell lung cancer. Genomics Proteomics Bioinformatics. 2014;12:198–209.
- Patel SP, Schwaederle M, Daniels GA, et al. Molecular inimitability amongst tumors: implications for precision cancer medicine in the age of personalized oncology. Oncotarget. 2015;6(32):32602–32609.
- Zhang J, Fujimoto J, Zhang J, et al. Intratumor heterogeneity (ITH) of lung adenocarcinomas defined by multiregion whole exome sequencing (WES). J Clin Oncol. 2014;32:5s. suppl; abstr 11032
- Jiang T, Ren S, Zhou C. Role of circulating-tumor DNA analysis in non-small cell lung cancer. Lung Cancer. 2015;90(2):128–134.
- Karachaliou N, Mayo-De Las Casas C, Queralt C, et al. Association of EGFR L858R Mutation in Circulating Free DNA With Survival in the EURTAC Trial. JAMA Oncol. 2015;1:149–157.
- Micke P, Edlund K, Holmberg L, et al. Gene copy number aberrations are associated with survival in histologic subgroups of non-small cell lung cancer. J Thorac Oncol. 2011;6:1833–1840.
- Syrigos KN, Vansteenkiste J, Parikh P, et al. Prognostic and predictive factors in a randomized phase III trial comparing cisplatin-pemetrexed versus cisplatin-gemcitabine in advanced non-small-cell lung cancer. Ann Oncol. 2010;21:556–561.
- Huang YT, Lin X, Liu Y, et al. Cigarette smoking increases copy number alterations in non small-cell lung cancer. Proc Natl Acad Sci U S A. 2011;108:16345–16350.
- Tekpli X, Landvik NE, Skaug V, et al. Functional effect of polymorphisms in 15q25 locus on CHRNA5 mRNA, bulky DNA adducts and TP53 mutations. Int J Cancer. 2013;132(8):1811–1820.
- Deben C, Deschoolmeester V, Lardon F, et al. TP53 and MDM2 genetic alterations in non-small cell lung cancer: Evaluating their prognostic and predictive value. Crit Rev Oncol Hematol 2015; pii: S1040-8428(15)30089-5. 10.1016/j.critrevonc.2015.11.019. [Epub ahead of print]
- Galvani E, Peters GJ, Giovannetti E, Thymidylate synthase inhibitors for non-small cell lung cancer. Expert Opin Investig Drugs. 2011;20:1343–1356.
- Chattopadhyay S, Moran RG, Goldman ID. Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol Cancer Ther. 2007;6:404–417.
- Giovannetti E, Lemos C, Tekle C, et al. Molecular mechanisms underlying the synergistic interaction of erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor, with the multitargeted antifolate pemetrexed in non-small-cell lung cancer cells. Mol. Pharmacol. 2008;73:1290–1300.
- Takezawa K, Okamoto I, Okamoto W, et al. Thymidylate synthase as a determinant of pemetrexed sensitivity in non-small cell lung cancer. Br J Cancer. 2011;104:1594–1601.
- Hanauske AR, Eismann U, Oberschmidt O, et al. In vitro chemosensitivity of freshly explanted tumor cells to pemetrexed is correlated with target gene expression. Invest New Drugs. 2007;25:417–423.
- Chen CY, Chang YL, Shih JY, et al. Thymidylate synthase and dihydrofolate reductase expression in non-small cell lung carcinoma: The association with treatment efficacy of pemetrexed. Lung Cancer. 2011;74:132–138.
- Sun JM, Han J, Ahn JS, et al. Significance of Thymidylate Synthase and Thyroid Transcription Factor 1 Expression in Patients with Nonsquamous Non-small Cell Lung Cancer Treated with Pemetrexed-Based Chemotherapy. J Thorac Oncol. 2011;6:1392–1399.
- Ceppi P, Volante M, Saviozzi S, et al. Squamous cell carcinoma of the lung compared with other histotypes shows higher messenger RNA and protein levels for thymidylate synthase. Cancer. 2006;107:1589–1596.
- Wang L, Wang R, Pan Y, et al. The pemetrexed-containing treatments in the non-small cell lung cancer is -/low thymidylate synthase expression better than +/high thymidylate synthase expression: a meta-analysis. BMC Cancer. 2014;14:205.
- Sun JM, Ahn JS, Jung SH, et al. Pemetrexed Plus Cisplatin Versus Gemcitabine Plus Cisplatin According to Thymidylate Synthase Expression in Nonsquamous Non-Small-Cell Lung Cancer: A Biomarker-Stratified Randomized Phase II Trial. J Clin Oncol. 2015;33:2450–2456.
- Novello S, Scagliotti G, Torri V, et al. International tailored chemotherapy adjuvant trial: ITACA trial. J Clin Oncol. 2011;29:e17514. suppl; abstr
- Liu J, Schmitz JC, Lin X, et al. Thymidylate synthase as a translational regulator of cellular gene expression. Biochim Biophys Acta. 2002;1587(2–3):174–182.
- Horie N, Aiba H, Oguro K, et al. Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5ʹ-terminal regulatory region of the human gene for thymidylate synthase. Cell Struct Funct. 1995;20:191–197.
- Wang X, Wang Y, Wang Y, et al. Association of thymidylate synthase gene 3ʹ-untranslated region polymorphism with sensitivity of non-small cell lung cancer to pemetrexed treatment: TS gene polymorphism and pemetrexed sensitivity in NSCLC. J Biomed Sci. 2013;25:20–25.
- Li WJ, Jiang H, Fang XJ, et al. Polymorphisms in thymidylate synthase and reduced folate carrier (SLC19A1) genes predict survival outcome in advanced non-small cell lung cancer patients treated with pemetrexed-based chemotherapy. Oncol Lett. 2013;5:1165–1170.
- Hu Q, Li X, Su C, et al. Correlation between thymidylate synthase gene polymorphisms and efficacy of pemetrexed in advanced non-small cell lung cancer. Exp Ther Med. 2012;4:1010–1016.
- Smit EF, Burgers SA, Biesma B, et al. Randomized phase II and pharmacogenetic study of pemetrexed compared with pemetrexed plus carboplatin in pretreated patients with advanced non-small-cell lung cancer. J Clin Oncol. 2009;27:2038–2045.
- Tiseo M, Giovannetti E, Tibaldi C, et al. Pharmacogenetic study of patients with advanced non-small cell lung cancer (NSCLC) treated with second-line pemetrexed or pemetrexed-carboplatin. Lung Cancer. 2012;78:92–99.
- Adjei AA, Mandrekar SJ, Dy GK, et al. Phase II trial of pemetrexed plus bevacizumab for second-line therapy of patients with advanced non-small-cell lung cancer: NCCTG and SWOG study N0426. J Clin Oncol. 2010;28:614–619.
- Adjei AA, Salavaggione OE, Mandrekar SJ, et al. Correlation between polymorphisms of the reduced folate carrier gene (SLC19A1) and survival after pemetrexed-based therapy in non-small cell lung cancer: a North Central Cancer Treatment Group-based exploratory study. J Thorac Oncol. 2010;5:1346–1353.
- Erdem L, Giovannetti E, Leon LG, et al. Polymorphisms to predict outcome to the tyrosine kinase inhibitors gefitinib, erlotinib, sorafenib and sunitinib. Curr Top Med Chem. 2012;12:1649–1659.
- Galvani E, Alfieri R, Giovannetti E, et al. Epidermal growth factor receptor tyrosine kinase inhibitors: current status and future perspectives in the development of novel irreversible inhibitors for the treatment of mutant non-small cell lung cancer. Curr Pharm Des. 2013;19:818–832.
- Politi K, Ayeni D, Lynch T. The Next Wave of EGFR Tyrosine Kinase Inhibitors Enter the Clinic. Cancer Cell. 2015;27:751–753.
- Nomura M, Shigematsu H, Li L, et al. Polymorphisms, mutations, and amplification of the EGFR gene in non-small cell lung cancers. PLoS Med. 2007;4:e125.
- Moriai T, Kobrin MS, Hope C, et al. A variant epidermal growth factor receptor exhibits altered type alpha trans- forming growth factor binding and transmembrane signaling. P Natl Acad Sci USA. 1994;91:10217–10221.
- Liu W, Innocenti F, Wu MH, et al. A functional common polymorphism in a Sp1 recognition site of the epidermal growth factor receptor gene promoter. Cancer Res. 2005;65:46–53.
- Liu G, Gurubhagavatula S, Zhou W, et al. Epidermal growth factor receptor polymorphisms and clinical outcomes in non-small-cell lung cancer patients treated with gefitinib. Pharmacogenomics J. 2008;8:129–138.
- Ichihara S, Toyooka S, Fujiwara Y, et al. The impact of epidermal growth factor receptor gene status on gefitinib-treated Japanese patients with non-small-cell lung cancer. Int J Cancer. 2007;120:239–247.
- Shitara M, Sasaki H, Yokota K, et al. Polymorphisms in intron 1 of the EGFR gene in non-small cell lung cancer patients. Exp Ther Med. 2012;4:785–789.
- Gregorc V, Hidalgo M, Spreafico A, et al. Germline polymorphisms in EGFR and survival in patients with lung cancer receiving gefitinib. Clin Pharmacol Ther. 2008;83:477–484.
- Gebhardt F, Bürger H, Brandt B. Modulation of EGFR gene tran- scription by secondary structures, a polymorphic repetitive se- quence and mutations–a link between genetics and epigenetics. Histol Histopathol. 2000;15:929–936.
- Amador ML, Oppenheimer D, Perea S, et al. An epidermal growth factor receptor intron 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer Res. 2004;64:9139–9143.
- Gebhardt F, Zänker KS, Brandt B. Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J Biol Chem. 1999;274:13176–13180.
- Buerger H, Gebhardt F, Schmidt H, et al. Length and loss of heterozygosity of an intron 1 polymorphic sequence of egfr is related to cytogenetic alterations and epithelial growth factor receptor expression. Cancer Res. 2000;60:854–857.
- Han S-W, Jeon YK, Lee KH, et al. Intron 1 CA dinucleotide repeat polymorphism and mutations of epidermal growth factor receptor and gefitinib responsiveness in non-small-cell lung cancer. Pharmacogenet Genom. 2007;17:313–319.
- Ma F, Sun T, Shi Y, et al. Polymorphisms of EGFR predict clinical outcome in advanced non-small-cell lung cancer patients treated with Gefitinib. Lung Cancer. 2009;66:114–119.
- Dubey S, Stephenson P, Levy DE, et al. EGFR dinucleotide repeat polymorphism as a prognostic indicator in non-small cell lung cancer. J Thorac Oncol. 2006;1:406–412.
- Nie Q, Wang Z, Zhang GC, et al. The epidermal growth factor receptor intron1 (CA) n microsatellite polymorphism is a potential predictor of treatment outcome in patients with advanced lung cancer treated with gefitinib. Eur J Pharmacol. 2007;570:175–181.
- Tiseo M, Rossi G, Capelletti M, et al. Predictors of gefitinib outcomes in advanced non-small cell lung cancer (NSCLC): study of a comprehensive panel of molecular markers. Lung Cancer. 2010;67:355–360.
- Brugger W, Triller N, Blasinska-Morawiec M, et al. Prospective molecular marker analyses of EGFR and KRAS from a randomized, placebo-controlled study of erlotinib maintenance therapy in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29:4113–4120.
- Lemos C, Kathmann I, Giovannetti E, et al. Impact of cellular folate status and epidermal growth factor receptor expression on BCRP/ABCG2-mediated resistance to gefitinib and erlotinib. Br J Cancer. 2009;100:1120–1127.
- Harris SL, Gil G, Robins H, et al. Detection of functional single-nucleotide polymorphisms that affect apoptosis. Proc Natl Acad Sci U S A. 2005;102(45):16297–16302.
- Emamian ES, Hall D, Birnbaum MJ, et al. Convergent evidence for impaired AKT1–GSK3b signaling in schizophrenia. Nat Genet. 2004;36:131–137.
- Giovannetti E, Zucali PA, Peters GJ, et al. Association of polymorphisms in AKT1 and EGFR with clinical outcome and toxicity in non-small cell lung cancer patients treated with gefitinib. Mol Cancer Ther. 2010;9:581–593.
- Avan A, Maftouh M, Avan A, et al. SNPs in PI3K-PTEN-mTOR and brain metastases in NSCLC–letter. Clin Cancer Res. 2014;20:3623–3624.
- Kim MJ, Kang HG, Lee SY, et al. AKT1 polymorphisms and survival of early stage non-small cell lung cancer. J Surg Oncol. 2012;105:167–174.
- Lemos C, Jansen G, Peters GJ. Drug transporters: recent advances concerning BCRP and tyrosine kinase inhibitors. Br J Cancer. 2008;98:857–862.
- Porcelli L, Lemos C, Peters GJ, et al. Intracellular trafficking of MDR transporters and relevance of SNPs. Curr Top Med Chem. 2009;9:197–208.
- Li J, Cusatis G, Brahmer J, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6:432–438.
- Lemos C, Giovannetti E, Zucali PA, et al. Impact of ABCG2 polymorphisms on the clinical outcome and toxicity of gefitinib in non-small-cell lung cancer patients. Pharmacogenomics. 2011;12:159–170.
- Peréz-Soler R, Saltz L. Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol. 2005;23:5235–5246.
- Huang CL, Yang CH, Yeh KH, et al. EGFR intron 1 dinucleotide repeat polymorphism is associated with the occurrence of skin rash with gefitinib treatment. Lung Cancer. 2009;64:346–351.
- Rudin CM, Liu W, Desai A, et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol. 2008;26:1119–1127.
- Morales-Espinosa D, García-Román S, Karachaliou N, et al. Pharmacogenomics in the treatment of lung cancer: an update. Pharmacogenomics. 2015;16(15):1751–1760.
- Bronte G, Rolfo C, Giovannetti E, et al. Are erlotinib and gefitinib interchangeable, opposite or complementary for non-small cell lung cancer treatment? Biological, pharmacological and clinical aspects. Crit Rev Oncol Hematol. 2014;89:300–313.
- Galvani E, Toffalorio F, Peters GJ, et al. Pharmacogenetics of non-small cell lung cancer (NSCLC): time to “work it out”? Curr Pharm Des. 2014;20:3863–3874.
- Kelly PJ, Stallard N, Whittaker JC. Statistical design and analysis of pharmacogenetic trials. Stat Med. 2005;24:1495–1508.
- Maitland ML, Ratain MJ, Cox NJ, Interpreting P values in pharmacogenetic studies: a call for process and perspective. J Clin Oncol. 2007;25:4513–4515.
- Kirchheiner J, Fuhr U, Brockmoller J. Pharmacogenetics-based therapeutic recommendations – ready for clinical practice. Nat Rev Drug Discov. 2005;4:639–647.
- Le Morvan V, Bellott R, Moisan F, et al. Relationships between genetic polymorphisms and anticancer drug cytotoxicity vis-à-vis the NCI-60 panel. Pharmacogenomics. 2006;7:843–852.
- Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607.
- Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575.
- Daly AK. Genome-wide association studies in pharmacogenomics. Nat Rev Genet. 2010;11:241–246.
- Wu X, Ye Y, Rosell R, et al., et al. Genome-wide association study of survival in non-small cell lung cancer patients receiving platinum-based chemotherapy. J Natl Cancer Inst. 2011;103:817–825.
- Guchelaar HJ, Gelderblom H, Van Der Straaten T, et al. Pharmacogenetics in the cancer clinic: from candidate gene studies to next-generation sequencing. Clin Pharmacol Ther. 2014;95:383–385.
- U.S. Food and Drug Administration. http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics. Accessed 11 January 2016.
- Giovannetti E, Erozenci A, Smit J, et al. Molecular mechanisms underlying the role of microRNAs (miRNAs) in anticancer drug resistance and implications for clinical practice. Crit Rev Oncol Hematol. 2012;81:103–122.
- Robles AI, Arai E, Mathé EA, et al. An Integrated Prognostic Classifier for Stage I Lung Adenocarcinoma Based on mRNA, microRNA, and DNA Methylation Biomarkers. J Thorac Oncol. 2015;10:1037–1048.
- Okayama H, Schetter AJ, Ishigame T, et al. The expression of four genes as a prognostic classifier for stage I lung adenocarcinoma in 12 independent cohorts. Cancer Epidemiol Biomarkers Prev. 2014;23:2884–2894.
- Karachaliou N, Mayo-de-Las-Casas C, Molina-Vila MA, et al. Real-time liquid biopsies become a reality in cancer treatment. Ann Transl Med. 2015;3:36.
- Rolfo C, Fanale D, Hong DS, et al. Impact of microRNAs in resistance to chemotherapy and novel targeted agents in non-small cell lung cancer. Curr Pharm Biotechnol. 2014;15:475–485.