Abstract
An increase in bodyweight is generally associated with an increased risk of excessive fat-related metabolic diseases (EFRMD), including Type 2 diabetes mellitus, hypertension and dyslipidemia. However, not all patients who are overweight have EFRMD, and not all patients with EFRMD are significantly overweight. The adipocentric paradigm provides the basis for a unifying, pathophysiological process whereby fat gain in susceptible patients leads to fat dysfunction (‘sick fat’), and wherein pathological abnormalities in fat function (adiposopathy) are more directly related to the onset of EFRMD than increases in fat mass (adiposity) alone. But just as worsening fat function worsens EFRMD, improved fat function improves EFRMD. Peroxisome proliferator-activated receptor-γ agonists increase the recruitment, proliferation and differentiation of preadipocytes (‘healthy fat’) and cause apoptosis of hypertrophic and dysfunctional (including visceral) adipocytes resulting in improved fat function and improved metabolic parameters associated with EFRMD. Weight loss interventions, such as a hypocaloric diet and physical exercise, in addition to agents such as orlistat, sibutramine and cannabinoid receptor antagonists, may have favorable effects upon fat storage (lipogenesis and fat distribution), nutrient metabolism (such as free fatty acids), favorable effects upon adipose tissue factors involved in metabolic processes and inflammation, and enhanced ‘cross-talk’ with other major organ systems. In some cases, weight loss therapeutic agents may even affect metabolic parameters and adipocyte function independently of weight loss alone, suggesting that the benefit of these agents in improving EFRMD may go beyond their efficacy in weight reduction. This review describes how adiposopathy interventions may affect fat function, and thus improve EFRMD.
Adiposopathy is defined as a pathological adipose tissue dysfunction that may be promoted and exacerbated by fat accumulation (adiposity) and sedentary lifestyle in genetically susceptible patients. In other words, while it is true that modifiable lifestyle choices clearly play a role in fat function and dysfunction, it is also true that the predisposition to dysfunctional fat often has a strong 'chance' genetic component which is not modifiable.
EFRMD: Excessive fat-related metabolic disease; FFA: Free fatty acids

Positive caloric balance leads to increased energy storage, which is first achieved through mild adipocyte hypertrophy. Afterwards, continued positive caloric balance may result in a predominance of further adipocyte hypertrophy in which adipose tissue may become dysfunctional or alternatively, a predominance of recruitment of new, functional adipocytes in which overall adipose tissue maintains normal metabolism. The accumulation of visceral fat (and other similar acting fat depots), even by recruitment, proliferation and differentiation, may result in pathological fat dysfunction manifested by an increased net release of free fatty acids and an increased proinflammatory response, which is further amplified by visceral fat hypertrophy.
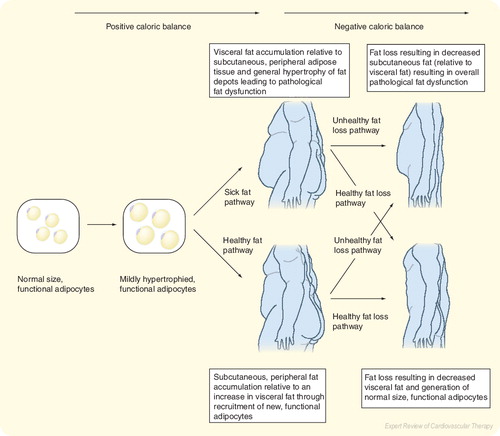
Adiposopathy is defined as pathological adipose tissue dysfunction that may be promoted and/or exacerbated by fat accumulation (adiposity) and sedentary lifestyle in genetically susceptible patients Citation[1]. During positive caloric balance, adiposopathy may be promoted by the following:
| • | Impaired adipogenesis; | ||||
| • | Pathogenic fat distribution; | ||||
| • | Impaired nutrient metabolism (such as an increase in the net release of free fatty acids [FFA]); | ||||
| • | Abnormal adipose tissue factor activity, otherwise responsible for favorable metabolic processes; | ||||
| • | Increased inflammation; | ||||
| • | Dysfunctional ‘cross-talk’ or pathological interactions with other body organ systems. | ||||
All of these abnormalities may contribute to excessive fat-related metabolic diseases (EFRMD), such as Type 2 diabetes mellitus (DM), hypertension and dyslipidemia Citation[301,302].
One approach towards improving fat function is to recruit and add more functional, or ‘healthy’ fat, and subtract or cause apoptosis of more dysfunctional ‘sick’ fat, such as through the use of peroxisome proliferator-activated receptor (PPAR)-γ agonists Citation[2,3]. Another approach to improve fat function in many overweight patients is through weight loss via a hypocaloric diet, increase in physical exercise and pharmaceutical agents that may not only result in weight loss, but may also prevent or lead to improvements in EFRMD Citation[4]. The mechanisms as to how adiposopathy causes EFRMD have been recently been reviewed Citation[3]. This report describes the effects of adiposopathy treatments on etiological mechanisms of pathological fat dysfunction.
Adipogenesis & adiposopathy
During positive caloric balance, energy must be stored, with the major energy storage organ being body fat. Stored body fat serves important physiological purposes, such as the release of lipids during fasting states, thus allowing for oxidation by skeletal muscle.
As this energy storage process takes place, adipocytes must optimally maintain function, because healthy fat function is essential to metabolic health. Storage of caloric balance is usually first accomplished through enlargement of fat cells (lipogenesis). After initial adipocyte hypertrophy, further fat storage occurs through signaling of the recruitment, proliferation and differentiation of ‘healthy’ adipocytes. The precise nature by which this adipogenic process is regulated is not always clearly defined. For example, depending upon the report, growth hormone has been described to both promote and inhibit adipogenesis. Furthermore, some factors may have differing effects within the adipogenic process, such as differing effects upon proliferation versus differentiation. In general, factors (not necessarily directly originating from adipose tissue) that may promote adipogenesis include hormones (estrogens, glucocorticoids, insulin, insulin growth factor [GF]-1, prolactin and thyroid hormone), proteins (neuropeptide Y), lipoproteins (very low density lipoproteins [VLDLs]) and lipids (endocannabinoids, circulating fatty acids, dietary saturated fats and prostaglandins). Factors not necessarily directly originating from adipose tissue that may impair adipogenesis include 5-hydroxytryptamine, androgens, catecholamines, various cytokines (such as tumor necrosis factor [TNF]-α) and interleukins (ILs), flavonoids, glucagon-like peptide-1, various GFs (such as epidermal GF, fibroblast GF, platelet-derived GF, transforming GF-β, and tumor GF-β), interferon, some prostaglandins and protein kinase C inhibitors Citation[3].
But adipogenesis is often most dependent upon specific adipocyte factors, including those that affect transcription, angiogenesis and extracellular matrix/cytoskeleton formation. Other adipocyte factors may be proadipogenic, antiadipogenic or in some cases both, depending upon whether the adipogenic process being affected is adipocyte proliferation or differentiation (Box 1).
If adipogenesis is impaired, then fat storage may occur through yet even further adipocyte hypertrophy, which if excessive, may cause adipose tissue to become dysfunctional or ‘sick’ Citation[3,5]. Thus, during times of positive caloric balance, the capacity for adipogenesis has significant influence on the predisposition to adiposopathy and, thus EFRMD.
The clinical implication of the adipogenesis and fat cell hypertrophy is dependent upon the inherent functionality of adipose tissue, which can be predicted by its location. Within subcutaneous, peripheral fat, increased proliferation and differentiation of adipocytes may maintain appropriate metabolic functions through increased capacity for energy storage during positive caloric balance. However, if adipogenesis is genetically limited or otherwise impaired, then adipocyte hypertrophy may occur, leading to dysfunction, or adiposopathy and thus promoting EFRMD Citation[3]. Conversely, if visceral fat (a more metabolically active fat depot) accumulates, even through proliferation and differentiation, then dysmetabolisms ensue leading to EFRMD, a process that is further amplified with visceral fat hypertrophy Citation[3].
Adiposopathy therapies
Regarding treatments that affect adipogenesis, weight loss downregulates adipogenic gene expression, including nuclear receptors, such as PPAR-γ receptors Citation[6]. It appears that it is fat mass loss, rather than the dietary fat:carbohydrate ratio, which is of most importance towards modifying the expression of adipogenic genes Citation[7]. One might presume that with negative caloric balance, additional energy storage is not required and, thus, adipogenesis is not needed. The effects of physical exercise alone and the effects of orlistat (a gastrointestinal lipase inhibitor) and sibutramine (a noradrenaline and serotonin reuptake inhibitor) Citation[4] on adipogenesis have not been well described in humans.
PPAR-γ agonists promote adipogenesis and improve adipocyte endocrine functions Citation[2,5,8] and, thus, serve as important therapeutic agents towards improving EFRMD, particularly Type 2 DM and dyslipidemia. Specifically, PPAR-γ agents cause proliferation and differentiation of preadipocytes into more functional fat cells in peripheral, subcutaneous fat, and apoptosis of differentiated large, more dysfunctional adipocytes in visceral and/or subcutaneous fat depots Citation[9].
Cannabinoid (CB)-1 receptor antagonists (in rodents) have been reported to reduce adipocyte fat content without adipocyte apoptosis Citation[10]. CB-1 blockade may also inhibit adipocyte proliferation Citation[11]. As with weight loss via a hypocaloric diet, this decrease in adipogenesis may be related to the negative caloric balance (through decreased appetite) promoted by CB-1 receptor antagonists or ‘cross-talk’ effects with other organ systems (such as liver, muscle, CNS/sympathetic nervous system or the gastrointestinal tract). CB-1 antagonism may also have direct effects upon adipose tissue, given that endo-CB receptors are found on adipocytes Citation[12]. Mechanistically, CB-1 antagonism may counteract CB-1’s otherwise stimulatory effects upon increasing intracytoplasmic cyclic adenosine monophosphate (cAMP) Citation[13] (increased cAMP is generally associated with increased adipogenesis) Citation[14]. Thus, CB-1 inhibition may directly improve adipose tissue functionality through a reduction in fat cell hypertrophy and maintenance of the existing number of functional adipocytes (limiting both proliferation and apoptosis). Clinically, improvement in fat function with CB-1 receptor antagonists is supported by improvements in glucose metabolism, triglycerides, high density lipoprotein (HDL) cholesterol, and reductions in the proportion of small, dense low density lipoprotein (LDL) particle levels, all found in human studies with the CB-1 receptor antagonists Citation[5].
In summary, a capacity to recruit, proliferate and differentiate adipocytes may allow adipose tissue to maintain normal metabolic functions during times of positive caloric balance. If excessive adipocyte hypertrophy results in adiposopathy, then therapies that promote the recruitment, proliferation and differentiation of subcutaneous, peripheral ‘healthy’ fat and apoptosis of hypertrophied ‘sick’ fat (such as PPAR-γ agents) lead to improvement in body metabolism and may serve to effectively treat EFRMD, even if total body fat is increased. Although not always so Citation[3], therapies that promote negative caloric balance often result in decreased adipocyte fat content, decreased need for adipogenic gene expression, improved fat function and improved metabolic parameters associated with adiposopathy and EFRMD .
Fat distribution & adiposopathy
Adipose tissue is a heterogeneous metabolic organ that can be segmented into different locations, which often reflect inherently different functionalities Citation[15]. Subcutaneous, peripheral fat (typically representing approximately 80% of total fat Citation[16]) includes truncal, gluteofemoral, mammary, inguinal (reported in animal studies) and subcutaneous abdominal fat. Visceral fat (typically approximately 20% of total fat) includes intraperitoneal (omental, mesenteric and umbilical), extraperitoneal (peripancreatic and perirenal), and intrapelvic (gonadal, such as epidydimal and urogenital) fat. Other fat depots include intraorgan fat (e.g., liver, muscle and bone) and periorgan fat (e.g., pericardial, perimuscular, perivascular, orbital and paraosseal). Subcutaneous, peripheral fat has been described as having the lowest level of metabolic activity. Visceral fat has direct access to the liver through the portal vein (which supplies 80% of the hepatic blood supply), and has been described as having the highest level of metabolic activity Citation[16,17] These different fat depots are genetically predetermined to have differences in the activity of various cellular receptors, differences in enzymatic processes involved with fat metabolism, and differences in the gene expression and production of bioactive molecules Citation[18,19]. Thus, owing to the fact that visceral fat accumulation generates the greatest metabolic activity through high basal lipolysis, increased sensitivity to catecholamines, and decreased sensitivity to insulin (all amplified by visceral fat hypertrophy) Citation[20] and because visceral fat accumulation and hypertrophy exhibits inflammatory activity (such as increased tumor-necrosis factor, IL-6, and plasminogen activator inhibitor type 1 with decreased adiponectin secretion) Citation[21], it is the accumulation and hypertrophy of visceral fat that is more associated with metabolic disease than overall fat Citation[22,23,24].
Other fat depots may differ in function as well. For example, abdominal subcutaneous fat may have metabolic activity between that of subcutaneous, peripheral fat and visceral fat. Excessive intraorgan (e.g., intrahepatic and intramuscular) fat promotes intraorgan dysmetabolisms, which may lead to EFRMD. Finally, periorgan fat (e.g., perirenal, orbital and pericardial) may also have metabolic activity between that of subcutaneous, peripheral fat and visceral fat, although it has been suggested that epicardial adipose tissue, at least with respect to adipose inflammatory responses, may be more functionally analogous to omental or visceral fat Citation[3,25].
Adiposopathy therapies
Some reports suggest that weight loss through diet and exercise decreases visceral fat quicker, and to a greater extent, than subcutaneous fat Citation[26,27] and that exercise promotes visceral fat loss, sometimes without changes in body mass index Citation[28]. If true, then this is possibly because visceral fat is more metabolically active and, thus, more susceptible to the physiological changes of weight loss. Findings such as these support the contention often stated in fitness magazines that a reduction in abdominal girth is among the first area of the body affected by weight loss. However, other scientific reports suggest that weight loss through bariatric surgery, a very low calorie diet (VLCD) and exercise all result in generally the same degree of fat loss in the visceral and subcutaneous, peripheral fat Citation[29,30], with no differences in visceral fat loss when comparing weight loss due to diet or exercise, or even compared with exercise without weight loss Citation[31]. Nonetheless, negative caloric balance in overweight patients, achieved through caloric restriction (with or without exercise), increases insulin sensitivity, which is mostly associated with decreases in total fat mass and visceral fat, relative to effects upon intrahepatic and intramyocellular lipids, or even fat cell size Citation[32]. With regard to the type of diet and fat distribution, it has been suggested that a low calorie/low carbohydrate diet may be more effective in reducing visceral fat than a low calorie/high carbohydrate diet, at least in obese subjects with Type 2 DM Citation[33].
With regard to drug therapy, PPAR-γ agents have generally been shown to decrease the visceral:subcutaneous fat ratio, with either a decrease or no change in visceral fat Citation[2,27]. This suggests the possibility that while apoptosis of hypertrophied, dysfunctional ‘sick’ fat cells with PPAR-γ agonists may contribute to favorable metabolic effects of these agents, it is the addition of ‘healthy’ subcutaneous, peripheral fat functionality that may play the greatest role in improving metabolic processes favorably affecting EFRMD.
Orlistat administration decreases both visceral and subcutaneous fat, with possibly more reduction of visceral fat Citation[34]. Weight loss with orlistat is the greatest contributor to improved insulin sensitivity observed with this agent Citation[34]. However, because orlistat impairs intestinal fat absorption, it has been suggested that in patients with Type 2 DM, orlistat may independently improve metabolic parameters, such as dyslipidemia, FFA levels and insulin sensitivity, beyond that of weight loss Citation[35]. Sibutramine administration has also been demonstrated to reduce waist circumference, waist-to-hip ratio and somewhat decrease visceral fat more than total abdominal fat and total subcutaneous fat (as assessed through imaging studies such as computerized tomography), which are all associated with favorable metabolic effects, such as upon fasting glucose and insulin levels Citation[36]. The CB-1 antagonist rimonabant has been demonstrated to reduce abdominal adiposity and the metabolic abnormalities associated with EFRMD Citation[37], likely owing to overall weight loss due to decreased CNS suppression of appetite but also potentially related to independent interactions with CB receptors directly associated with adipocytes Citation[38], or possibly enhanced ‘cross-talk’ with other organ systems affected by the endo-CB system.
In summary, therapies that decrease visceral fat are associated with improvements in metabolic health. Even if visceral fat remains constant, therapies that reduce the visceral:subcutaneous, peripheral fat ratio (through an addition of in ‘healthy’ subcutaneous, peripheral fat) may improve fat function, and may be effective treatment for EFRMD, even if overall fat is increased. Thus, while much attention has been given to the pathological consequences of increased visceral fat with respect to metabolic health, it should be understood that pathology of subcutaneous, peripheral fat may also have adverse metabolic health consequences Citation[3], with the implication that a therapeutic objective towards treating adiposopathy is to not only improve visceral fat function but total fat function as well.
Nutrient metabolism & adiposopathy
If caloric intake exceeds adipose tissue’s ability to appropriately store triglycerides (as may occur with limitations to or impairment of, recruitment, proliferation and differentiation of new fat cells and/or hypertrophy of existing fat cells), then a net release of FFAs into the circulation may occur Citation[39], which may be ‘lipotoxic’ to other body organs. It is the adiposopathic net increased release of FFAs which is the adipose tissue nutrient metabolic abnormality that most directly links excessive body fat to EFRMD Citation[1,2].
Approximately 80% of adipose tissue weight is lipid, with over 90% of lipids being stored triglycerides. FFAs (and glycerol) are generated when stored triglycerides undergo hydrolysis by intra-adipocyte lipases, and FFAs are quantitatively the major secretory product from adipose tissue Citation[40]. The excessive release or impaired uptake of FFA from dysfunctional adipocytes may result in a net increase in circulating FFAs resulting in chronic ectopic fat storage that promotes EFRMD such as Type 2 DM due to lipotoxicity to muscle and liver (contributing to insulin resistance) and pancreas (contributing to insulinopenia) Citation[41]. Increased FFA levels are also an independent risk factor for hypertension Citation[42], likely as the result of FFA-induced insulin resistance (which is associated with increased blood pressure), impairment of endothelium-dependent vasodilation and other microvascular dysfunctions leading to hypertension Citation[43,44]. Finally, elevated fasting or postprandial FFAs contribute to the typical dyslipidemia found with the ‘metabolic syndrome’, which includes hypertriglyceridemia, low HDL-cholesterol levels, and abnormalities of lipoprotein particle size and subclass distribution, such as small, dense low LDL particles Citation[3,45].
FFA metabolism is largely dependent upon the balance between intracellular adipocyte hormone-sensitive lipase (HSL) (which hydrolysis intra-adipocyte triglycerides) and the adipocyte secreted lipoprotein lipase (LPL) (which hydrolyzes extra-adipocyte triglycerides). The activity of HSL is increased with β-adrenergic stimulation and decreased with insulin, while the activity of LPL is decreased with β-adrenergic activity and increased with insulin.
During fasting, insulin levels are decreased and, thus, HSL activity is increased and LPL activity is decreased. The increase in intra-adipocyte triglyceride hydrolysis leads to increased circulating FFA, which normally undergo uptake in muscle as the primary fuel during prolonged fasting. Glycerol and FFAs also undergo uptake in the liver for promotion of hepatic gluconeogenesis and ketogenesis Citation[46].
After feeding, postprandial insulin levels are increased, decreasing HSL, and increasing LPL activity. Over 90% of FFAs become trapped by adipocytes, with lower postprandial circulating FFA levels compared with fasting FFA levels Citation[46]. However, with insulin resistance, the response of both HSL and LPL is impaired, with diminished suppression of intra-adipocyte triglyceride hydrolysis, decreased extra-adipocyte triglyceride hydrolysis, and an increase in postprandial circulating FFA and triglyceride levels Citation[3].
Adiposopathy therapies
Acutely, during starvation or abrupt weight loss through a VLCD, FFA net release is increased Citation[47] owing to the need for alternative energy extraction and mobilization from triglyceride stores in adipose tissue and VLDL Citation[48]. However, more gradual, long-term weight loss (6 to >12 months) through diet may actually decrease FFA blood levels Citation[49,50], presumably by improved fat function and energy storage capability, which would be expected to have favorable effects upon glucose metabolism. Endurance exercise also decreases FFA levels during and after and training Citation[51]. The reduction in FFA levels with gradual, long-term weight loss through diet and exercise is independently associated with increased insulin sensitivity and decreased vascular inflammation Citation[52], which might be expected to also improve EFRMD such as blood pressure. Finally, the improvements in FFA metabolism is a contributing reason why hypocaloric diets and physical exercise often decrease cholesterol and triglyceride levels, increase HDL-cholesterol levels Citation[53–55], and improve LDL and HDL lipoprotein particle sizes Citation[56,57]. PPAR-γ agonists decrease circulating FFAs Citation[58,59], as do weight-loss therapies, such as orlistat Citation[35,60], possibly sibutramine Citation[61,62] and CB-1 receptor antagonists Citation[63].
In summary, positive caloric balance may result in excessive adipocyte hypertrophy due to limitations or impairment of recruitment and proliferation of new fat cells. Excessive fat cell size decreases the responsiveness of adipose tissue to insulin and in general, adipocyte hypertrophy is more closely linked to metabolic abnormalities than is an increase in total body fat Citation[5]. Excessive fat cell hypertrophy results in abnormalities in nutrient metabolism, such as an excessive increase in the net release of FFA, and is an important adiposopathatic process wherein storage of further energy influx is inadequate, and adipocytes have a resistance or inability to store triglycerides beyond some maximal amount Citation[64]. Increased circulating FFAs are lipotoxic to peripheral tissues, and promote EFRMD. Given that increased circulating FFAs significantly contribute to dysmetabolism, it is not surprising that therapies that decrease FFAs are associated with improvements in EFRMD.
Adipose tissue factors (inflammatory & noninflammatory)
Adverse, pathological fat function associated with adiposopathy can result in increased or decreased production and/or activity of adipose tissue factors, which may result in a multiple adipose tissue dysmetabolisms, including proinflammatory responses, which all may contribute to EFRMD Citation[3]. Adipocyte factors include adipokines (bioactive secretary proteins such as cytokines, GFs, enzymes and peptide hormones) and nonadipokine cellular proteins, fatty acids, prostaglandins, lipids, steroid hormones and other essential cellular factors, similar to what is found in many body tissues. Adipocyte factors are active in adipogenesis, including extracellular matrix/cytoskeleton construct (formation, protease degradation and reformation of the surrounding adipose tissue extracellular matrix is necessary for maturation of adipocytes) (Box 1). Adipocyte factors are also important in many other body functions, including glucose and lipid metabolism, vascular tone, immunity/inflammation, steroid hormone production, angiogenesis and coagulation/fibrinolysis (Box 2).
Regarding inflammation, adiposopathy may contribute to EFRMD by the increased production of inflammatory factors and decreased production of anti-inflammatory factors. Proinflammatory adipose tissue factors include:
| • | Factors with cytokine activity (adipsin, IL-1B, -6, -8, -17D, -18, leptin, monocyte chemoattractant protein-1, macrophage migration inhibitory factor, resistin, TNF-α and regulated on activation, normal T-cell expressed and secreted (RANTES) chemokine); | ||||
| • | Acute phase response proteins (α-1 acid glycoprotein, ceruloplasmin, C-reactive protein, haptoglobin, IL-1 receptor antagonist, lipocalins, metallothionein, pentraxin-3, plasminogen activator inhibitor-1, and serum amyloid A); | ||||
| • | Proteins of the alternative complement system (adipsin, acylation-stimulating protein [ASP], complement C3 and B); | ||||
| • | Chemotactic/chemoattractants for immune cells (eotaxin, interferon inducible protein, monocyte chemoattractant protein-1, macrophage colony simulating factor, macrophage migration inhibitory factor, RANTES, stromal derived factor-1, vascular adhesion protein, and vascular cell adhesion molecule-1); | ||||
| • | Eicosanoids/prostaglandins, such as prostaglandin E2. | ||||
Adipocyte factors that may have anti-inflammatory effects include adiponectin, annexin-1, IL-6 and -10, transforming GF-β, bone morphogenic factor and nitric oxide.
Adiposopathy therapies
Weight loss in overweight and obese patients through a hypocaloric diet and increased physical exercise may favorably alter adipocyte factors and clinically improve glucose metabolism and Type 2 DM Citation[65–68], and hypertension Citation[69,70] as does weight loss in obese patients through bariatric surgery Citation[71]. Weight loss in overweight and obese patients through hypocaloric diet and increased physical exercise also improves dyslipidemia Citation[67,72,73]. With regard to diet, it may be the loss of fat mass, rather than the ratio of fat:carbohydrate that is of most importance towards modifying expression of adipocyte factor genes Citation[7].
Through their favorable effects upon adipocyte factors, therapeutic interventions may improve EFRMD such as Type 2 DM, hypertension and dyslipidemia. The number of the most extensively described human genes in adipose tissue number at least 8500 Citation[74]. Given that the dysfunction of many of these gene products may contribute to EFRMD, it would be impossible to discuss the effects of therapeutic intervention on every conceivable adipocyte factor. Furthermore, the effects of therapeutic interventions on many adipocyte factors known to contribute to EFRMD are unknown. But for illustrative purposes, the following is a brief discussion of the effects of weight loss through diet and physical exercise on selected adipocyte factors as it relates to Type 2 DM, hypertension and dyslipidemia. list these effects, as well as the effects of comparable drug treatments (which are not discussed in detail).
Diet & exercise & specific adipocyte factors
11-β-hydroxysteroid dehydrogenase type 1 (11 HSD-1) is an adipocyte enzyme that catalyses inactive cortisone to active cortisol. Excessive body fat is associated with increased expression of 11HSD-1 Citation[3,75–77] and increased cortisol levels may contribute to impaired glucose metabolism. This association with excessive body fat appears to be tissue specific with increased 11HSD-1 activity in adipose tissue and skeletal muscle (contributing to insulin resistance), and decreased 11HSD-1 activity in the liver Citation[78]. Weight loss through diet does not change Citation[79], or may actually increase total body 11HSD-1 activity, with increased cortisol:cortisone ratio Citation[77]. But again, interventions that affect 11HSD-1 may be tissue specific, in that physical exercise may result in decreased 11HSD-1 protein expression with decreased glucocorticoid action in skeletal muscle, while increased 11HSD-1 activity may occur in visceral fat, even while total visceral fat mass is decreased Citation[80].
The decrease in muscle 11HSD-1 activity may help to decrease insulin resistance, despite no change or increase in overall 11HSD-1. The extent that alterations in 11HSD-1 may worsen or improve high blood pressure Citation[81–83], may not be as strongly linked to hypertension in humans as in mice Citation[84]. Finally, increased 11HSD-1 activity has been associated with the lipid pattern often seen with the metabolic syndrome Citation[81,83–85]. But even if weight loss is associated with further increases in adipocyte 11HSD-1 activity, cholesterol levels are nonetheless decreased Citation[76].
Acylation-stimulating protein
ASP is generally increased with excessive body fat Citation[3] and is an adipocyte factor that functions to enhance short-term energy clearance and fat storage through clearance of circulatory FFAs, increased triglyceride synthesis and decreased FFA release in adipocytes Citation[86,87]. In fact, ASP might be considered to be the most potent stimulant of adipocyte triglyceride synthesis Citation[18]. ASP production is stimulated by insulin and triglyceride containing lipoproteins (chylomicron/VLDL). Adipocyte triglyceride synthesis is facilitated by ASP-induced increases in insulin secretion, increased glucose uptake, a decrease in HSL activity, an increase in LPL activity, and an increase in diacylglycerol acyltransferase (DGAT) activity, which is a key microsomal enzyme involved in triglyceride synthesis Citation[87–90]. ASP may also enhance differentiation of preadipocytes, as well as have effects upon cytokine and pituitary hormone secretions which potentially affect glucose metabolism Citation[91].
Impaired caloric absorption/intake is among the mechanisms of action of weight loss with bariatric gastric bypass, and this procedure in obese patients has been shown to decrease ASP levels Citation[92]. ASP levels may also be decreased with moderate weight loss through diet Citation[93]. With regard to exercise, ASP levels may be significantly reduced after short-term (2 weeks) endurance training Citation[94]. But if it is an increase in ASP activity that is associated with improved triglyceride storage, then how is the decrease in ASP activity induced by weight loss associated with favorable metabolic results? While it is probable that in many cases, improvement in the production of adipocyte factors is reflective of improved fat health, which directly improves EFRMD, it is also probable that in other cases alterations in adipocyte factors, such as a reduction in ASP levels due to diet and/or exercise, are simply a normal physiological response to other metabolic changes associated with weight loss.
Adiponectin
Adiponectin is an adipocyte factor that decreases with excessive body fat. Adiponectin functions to increase insulin sensitivity and decrease glucose levels Citation[87,95]. Dramatic weight loss through bariatric surgery increases adiponectin levels Citation[96], as does modest and gradual fat loss Citation[97,98]. In contrast, weight loss through abrupt fasting or implementation of VLCDs or through high intensity exercise (even with high caloric diet) may not increase adiponectin levels Citation[99–101]. Thus, weight loss through bariatric surgery may have physiological differences compared with weight loss through diet in affecting adipocyte function, and an increase in adiponectin levels may not be required for the observed improvements in glucose metabolism with acute weight loss through hypocaloric diets Citation[102]. Similarly, adiponectin levels may not be increased with exercise alone, despite increased insulin sensitivity, again suggesting that the favorable effects of exercise upon glucose metabolism may be through mechanisms other than effects upon adiponectin levels Citation[103].
As with potential improvements in glucose metabolism with increased adiponectin levels sometimes found with gradual weight loss, increased adiponectin production may also reverse the adverse metabolic processes that may lead to high blood pressure (such as adverse effects upon endothelial and smooth muscle cells) Citation[104,105]. The effects of weight loss on adiponectin levels may account for the nonsignificant increases in adiponectin and slight decreases in blood pressure achieved in some studies of weight loss through diet and exercise Citation[106].
With regard to dyslipidemia, decreased adiponectin levels are associated with increased triglyceride levels, and reduced HDL-cholesterol levels Citation[107]. Despite the variable association of adiponectin levels with weight loss through a hypocaloric diet and physical exercise, increased adiponectin levels have been associated with increased HDL-cholesterol, with perhaps some improvement in other lipid parameters of unclear significance Citation[106].
Adipsin
Adipsin is positively correlated with excessive body fat Citation[108] and insulin resistance, and participates in the formation of ASP (described above) Citation[48]. In addition to glucose effects, adipsin and ASP are involved in the adipocyte esterification of FFAs into triglycerides Citation[109]. Increased adipsin increases postprandial triglyceride clearance, while decreased adipsin may decrease postprandial triglyceride clearance Citation[88].
While weight loss through starvation may decrease various complement proteins of the alternative pathway (including adipsin, otherwise known as complement factor D) Citation[110], weight reduction through diet in weeks to months may not significantly decrease adipsin levels Citation[111]. The effects of exercise on adipsin levels or activity have not been well described.
Angiotensinogen
Angiotensinogen and possibly other components of the renin angiotensin system (angiotensin I, angiotensin II, angiotensin converting enzyme [ACE], renin, cathepsin D, cathepsin G and chymase) are all adipocyte factors, increased in overweight or obese patients, that contribute to adiposopathy by inhibiting the differentiation of adipocyte precursors, impairing adipocyte reserve, promoting adipocyte hypertrophy, inhibiting pancreatic insulin release and generally contributing to metabolic processes that adversely affect glucose metabolism Citation[112]. Modest weight loss in obese patients through dietary intervention may decrease angiotensinogen levels, decrease RAS factors and interestingly, these findings are highly correlated to a reduction in waist circumference Citation[113], which may reflect a reduction of visceral fat. As noted before, a reduction in visceral fat is associated with improved glucose metabolism Citation[1].
With regard to physical exercise, acute exercise may actually enhance RAS in the kidneys, induce renal vasoconstriction that reduces renal blood flow and, thus, redistribute blood to active muscle Citation[114]. Alternatively, chronic exercise may help reduce blood pressure in some patients, with the response at least possibly related to the polymorphism of the angiotensinogen gene Citation[115]. If chronic physical exercise does reduce RAS enzyme expression in some patients, then this may counteract the inhibition of adipocyte differentiation by angiotensinogen/ angiotensin II that may contribute to insulin resistance Citation[112], and therefore would be expected to improve glucose metabolism.
Similarly, because the RAS is integral to blood pressure, an increase in RAS activity (often found in overweight or obese patients) may be an important contributor to hypertension Citation[116–119]. Weight reduction, especially with waist circumference decline, has been demonstrated to reduce adipose tissue RAS enzyme levels, which is accompanied by a significant reduction in blood pressure Citation[113].
Autotaxin
Autotaxin catalyzes lysophosphatidic acid synthesis, which activates preadipocyte proliferation Citation[120] and, thus, may improve adipose tissue metabolic functions such as increased glucose sensitivity. Possibly as a corrective physiological response to other metabolic processes that may promote insulin resistance, impaired glucose tolerance, and/or Type 2 DM Citation[121], as well as increased TNF-α (which may promote insulin resistance), autotaxin expression may be increased with excessive body fat. The effects of dietary weight loss and physical exercise upon adipocyte autotaxin secretion have not been well described.
Ceramide
Ceramide is a signal mediator sphingolipid that accumulates intracellularly with the accumulation of excessive FFAs in adipose and other tissues Citation[122], as well as through increased inflammatory factors, such as TNF-α. Ceramide is involved in apoptosis, cell growth, senescence, and cell cycle control Citation[123], and may induce insulin resistance in adipose tissue and peripheral tissues such as muscle through inhibition of insulin signaling Citation[122,124–127]. Furthermore, chronically increased FFAs associated with adiposopathy may increase ceramide synthesis in other organs, such as the pancreas, resulting in increased β-cell apoptosis (programmed death), which further worsens glucose metabolism Citation[128].
Although not reported in human clinical trials, it is presumed that a decrease in FFAs, as might occur with dietary interventions that decrease both pre- and postprandial FFAs, might decrease the formation of ceramide, and thus improve adipocyte signaling functions such as improved glucose metabolism. While the effects of exercise on ceramide levels have been described in skeletal muscle Citation[129], less is known about the effects of exercise on adipocyte ceramide levels. This illustrates that many of the factors associated with adipose tissue that contribute to EFRMD are also produced by other organs. Thus, the effects of interventions such as weight loss and physical exercise may simultaneously affect similar factors associated with different organs and may affect similar metabolic processes that concomitantly improve EFRMD
Cholesteryl ester transfer protein.
Cholesteryl ester transfer protein (CETP) is increased with excessive body fat, is expressed in preadipocytes and is present throughout adipocyte differentiation Citation[130]. Increased CETP blood levels may contribute to hypertriglyceridemia, low HDL-cholesterol levels and, small dense LDL particles, often found with the metabolic syndrome Citation[131]. Reduction in body weight through surgical intervention reduces CETP levels, and improves LDL particle size Citation[132]. Exercise may decrease CETP as well, which, in turn, is associated with reductions in triglyceride and increases in HDL-cholesterol levels Citation[133].
Hormone-sensitive lipase
HSL is generally considered the principal regulator of lipolysis within adipose tissue and is the rate-limiting step for the release of FFAs from triglycerides stored in adipose tissue into the circulation Citation[134]. Although its expression may be decreased with excessive body fat Citation[135], the increase in lipolysis found in visceral versus peripheral fat (which may then lead to EFRMD) is likely to be due to sufficient HSL activity coupled with increased expression of β-adrenergic receptors, impaired inhibition of lipolysis by insulin and the effects of perilipin Citation[136].
During starvation, VLCDs, low-calorie diets and acute exercise Citation[7,137], HSL activity and, thus, lipolysis, is increased, particularly in visceral fat Citation[138]. Conversely, endurance training may decrease HSL activity and, thus, might reduce overall net adipocyte FFA release and improve metabolic processes that might otherwise promote EFRMD Citation[139]. This is one example supporting the fact that weight reduction towards the goal of improving EFRMD is best achieved through a combination of both hypocaloric diets (that result in weight loss) and increased physical exercise (that may decrease circulating FFAs).
Interleukin-6
IL-6 is generally increased in overweight or obese patients and reduces peripheral insulin receptor signaling components, suppresses cytokine signaling, inhibits adipogenesis, decreases adiponectin and decreases CNS IL-6 which are all associated with excessive body fat and associated metabolic abnormalities Citation[87], including impaired glucose tolerance and insulin resistance. IL-6 may also promote angiotensin II-sodium hypertension Citation[140], and may increase vascular resistance through other mechanisms Citation[141,142]. Diet and exercise may decrease adipocyte low grade inflammation in obese patients, as reflected by decreased IL-6 expression Citation[68,143], which may improve glucose metabolism and decrease blood pressure Citation[52].
Insulin-like growth factor-1
Insulin-like GF (IGF)-1 is an adipose-derived factor Citation[144,145], which is increased with excessive body fat, is regulated by IGF-binding proteins, and stimulates differentiation of pre-adipocytes Citation[146]. IGF-1 mediates some of the actions of growth hormone but does not mediate all growth hormone effects. For example, IGF-1 increases insulin sensitivity, while growth hormone itself decreases insulin sensitivity Citation[147].
Bariatric surgery may increase IGF-1 Citation[148], as does the weight loss through a hypocaloric diets, which are associated with decreases in insulin concentrations Citation[149]. Increased physical exercise added to a hypocaloric diet in obese patients with Type 2 DM may not result in any significant change in IGF-1 Citation[150]. However, it should be noted that IGF-1 is an example of an adipocyte factor secreted by a number of tissues. Thus, the relative importance of adipocyte secretion of IGF-1 and the impact towards the development of EFRMD is unclear.
Leptin
Leptin transfers information from adipose tissue to the CNS regarding the sufficiency of energy stores and leptin levels generally correlate to the amount of body fat. Leptin has physiological functions that promote a reduction in body fat Citation[4] but also has close metabolic ties to glucose metabolism Citation[151]. For example, in overweight or obese patients, leptin may counteract metabolic processes leading to EFRMD (e.g., Type 2 DM) by promoting increased peripheral insulin sensitivity, increased skeletal muscle glucose uptake and oxidation, suppression of hepatic glucose output, and decreased peripheral organ lipotoxicity that may otherwise lead to insulin insensitivity or resistance Citation[152]. While some reports suggest that leptin may increase insulin secretion from the pancreatic islet cells Citation[153], most other reports have demonstrated that leptin decreases insulin secretion Citation[154,155], which is an effect that may be dependent upon dose and other parameters Citation[154,156].
Hypocaloric diets and chronic exercise reduce leptin levels Citation[7,143,157]. Given the effects of leptin that would seem to promote weight loss and improved multiple metabolic processes, reduction in leptin levels through a hypocaloric diet and physical exercise may not be advantageous towards continued weight loss or even maintenance of weight loss, nor would a reduction in leptin levels be expected to promote continued improvement in metabolic parameters. In fact, a weight-reduced state might be analogous to leptin insufficiency. Since the practical effect of leptin in humans is primarily to signal energy adequacy, lower levels of leptin are likely to be of more importance towards stimulating feeding and decreasing energy expenditure than any effects higher leptin levels may have on reducing excessive body fat Citation[3]. Thus, one might consider the post-weight loss reduction of leptin as disadvantageous towards maintaining weight loss. Not surprisingly, administration of leptin after weight loss may prevent weight loss regain through increased energy expenditure, increased skeletal muscle work efficiency, increased sympathetic nervous system tone, and improved thyroid levels Citation[158].
While the increased leptin levels associated with excessive body fat may be at least potentially beneficial with regard to glucose (and lipid metabolism), the same might not apply to blood pressure. Acutely administered leptin has neutral effects upon blood pressure, likely because it stimulates processes that promote decreased blood pressure, such has naturesis and nitric oxide production (resulting in vasorelaxation) Citation[117], that may balance processes that increase blood pressure, such as leptin-promoted increases in sympathetic adrenergic activity Citation[105,159]. However, after chronic exposure to high leptin levels, blood pressure may be increased due to impairment or downregulation of the blood pressure lowering effects, and persistent increased sympathetic nervous system activity, increased oxidative stress, increased endothelin, enhanced renal sodium reabsorption, and possibly an eventual decrease in nitric oxide Citation[117,159]. Weight reduction through a hypocaloric diet and chronic exercise may decrease blood pressure, which in part, may be due to a reduction in leptin levels, which in turn, may decrease leptin-mediated sympathetic nervous system activity Citation[160].
With specific regard to dyslipidemia, leptin replacement therapy in leptin-deficient lipodystrophy decreases triglyceride levels Citation[161]. Interestingly, hyper-triglyceridemia may impair leptin’s transport across the blood–brain barrier, perhaps resulting in ‘leptin resistance’ Citation[162], which is a term that has been used to describe a reduction in leptin’s CNS signaling. Alternatively, a decrease in CNS leptin signaling can occur due to reductions in leptin blood levels caused by weight loss through a hypocaloric diet and increased physical exercise Citation[163]. As noted before, weight loss has the potential to reduce leptin levels which (by a compensatory response), may facilitate increased food intake, decreased energy expenditure, worsen adiposity, adiposopathy and theoretically promote dyslipidemia. However, given that the addition of physical exercise to hypocaloric diet improves acute and longer-term fat weight loss Citation[164], and given that increased physical activity decreases leptin levels, it is possible that irrespective of its effects upon leptin blood levels, increased physical activity may not only help decrease body fat, but may also help decrease ‘leptin resistance’, increase leptin CNS sensitivity Citation[165], and therefore help decrease body fat. This is yet another example how increased physical exercise may be complementary to hypocaloric diets in maintaining fat weight loss.
Lipin
Lipin is increased with increased body fat, and promotes fat accumulation and increases insulin sensitivity. Increased lipin improves glucose metabolism Citation[166], while a deficiency of lipin impedes the differentiation of adipocytes and worsens insulin resistance Citation[166]. The effects of a weight reduction diet and physical exercise on lipin have not been well described.
Lipoprotein lipase
LPL is increased with excessive body fat and human adipocytes rely upon LPL mediated hydrolysis of circulating triglycerides Citation[167], which results in the creation of FFAs which may undergo uptake by adipocytes for re-esterification. As noted before, excessive body fat is often associated with increased FFAs, resulting in increased VLDL production. An impaired increase/relative decrease in adipocyte LPL activity may partially account for impaired postprandial triglyceride clearance found in patients with increased visceral fat Citation[168]. Weight reduction through a VLCD may not affect, or perhaps further increase LPL levels Citation[196,170]. More gradual weight loss through a hypocaloric diet accompanied by an increase in physical exercise may also increase LPL activity Citation[171] and improve dyslipidemia Citation[172] not unlike that of fibrates Citation[173], although gender differences in lipoprotein lipase expression may exist Citation[174].
Lysophospholipids
Lysophospholipids, such as lysophosphatidic acid, are increased with excessive body fat and regulate cellular responses including proliferation, survival, motility, ion flux and secretion Citation[120]. Their secretion may be increased by autotaxin, which activates preadipocyte proliferation Citation[120,175], and which may provide additional adipose functionality and thus improve glucose metabolism. The effect of a weight reduction diet and physical exercise on lysophospholipids has not been well described.
Perilipin
Perilipin coats the lipid-storage droplets in adipocytes, and protects against HSL activity Citation[176,177], which might otherwise increase the release of FFAs Citation[178], lead to potential lipotoxicity Citation[2], and thus insulin resistance and Type 2 DM Citation[179]. The mass of perilipin per fat cell is constant between lean versus obese patients, and does not increase with increased fat volume Citation[178]. Thus, because perilipid mass remains constant, the larger the fat droplet (as might occur with excessive body fat), the more exposed the droplet may be to adipocyte lipases, and the greater the net release of FFAs. Genetic variants in perilipin in humans may influence the degree of weight loss achieved through hypocaloric diets Citation[180]. In summary, the amount and activity of perilipin is more likely to be more dependent upon genetic inheritance, lipid droplet size and other variables, rather than directly influenced by a hypocaloric diet and physical exercise.
Phosphoenolpyruvate carboxykinase
Phosphoenolpyruvate carboxykinase (PEPCK) activity increases with excessive body fat and functions to increase adipocyte size and fat mass by re-esterifying FFAs within fat cells Citation[181]. A deficiency of adipose PEPCK may increase FFA release, increase lipotoxicity, and increase insulin resistance and the risk of Type 2 DM Citation[182]. The relationship between PEPCK and weight reduction through diet and exercise has not been well described in humans.
Phospholipid transfer protein
Phospholipid transfer protein (PLTP) is increased with excessive body fat, and catalyses the transfer of phospholipids from triglyceride-rich lipoproteins to HDL particles Citation[183] and is increased with excessive body fat Citation[184]. The dyslipidemia often found with the metabolic syndrome is associated with increased PLTP activity, and thus an increased risk of atherosclerotic coronary heart disease (CHD) – although PLTP’s effects upon HDL-cholesterol levels and size are sometimes unclear Citation[185]. Pronounced weight loss after gastric-banding surgery may result in a significant decrease in PLTP activity and an increase of high-density particle size Citation[186]. However, dietary interventions such as Therapeutic Lifestyle Change Step 2 diet Citation[187] may have no effect upon PLTP levels, even while LDL-cholesterol, and HDL-cholesterol levels are reduced Citation[188]. The effect of exercise on adipose PLTP has not been well described.
Plasminogen activator inhibitor-1
Plasminogen activator inhibitor (PAI)-1 increases with excessive body fat, particularly with increased visceral fat and is positively correlated with Type 2 DM Citation[189,190]. Although not always consistent in the medical literature Citation[191], increased PAI-1 activity may adversely decrease adipocyte differentiation, while decreased PAI-1 activity may promote glucose uptake and adipocyte differentiation through increased PPAR-γ expression Citation[192]. Weight reduction through dietary and physical exercise intervention may reduce PAI-1 levels in obese patients, and reduce glucose intolerance Citation[193].
Resistin
Resistin increases with excessive body fat (especially visceral fat) and may impair glucose tolerance, decrease insulin mediated glucose uptake, and increase hepatic glucose production. However, the inconsistency of these findings suggests that resistin’s effects upon glucose metabolism remains undetermined Citation[48,194]. Weight loss through hypocaloric diets and increased physical exercise may not change resistin levels Citation[143].
Retinol binding protein
Retinol binding protein (RBP) is a transport protein for retinol which may increase with excessive body fat, and whose increased levels are associated with insulin resistance and Type 2 DM, at least partially due to a decrease in glucose transporters. Decreases in RBP improves insulin sensitivity Citation[195]. Hypocaloric diets do not appear to change RBP levels, which may be more related to protein intake Citation[196]. The effects of exercise on RBP have not been well described.
Sex hormones
Sex hormones are often altered with visceral obesity, with less testosterone and more estrogens in obese men, and more androgens in overweight women Citation[3]. Increased androgens lower HDL-cholesterol, increase LDL-cholesterol levels and decrease triglyceride levels, while increased estrogens raise HDL-cholesterol, lower LDL-cholesterol, and increase triglyceride levels Citation[197]. Weight loss through a hypocaloric diet (including behavior modification) increases testosterone levels in obese men Citation[198,199]. Conversely, estrogen levels may decrease Citation[200], or may not change with dramatic weight loss Citation[201,202]. Weight loss in obese women with polycystic ovarian syndrome through bariatric surgery decreases androgens Citation[203]. Weight loss through a hypocaloric diet Citation[149] or increased physical exercise in obese women with polycystic ovarian syndrome, on balance, also decreases androgens Citation[204]. It is unclear how much these changes in sex hormones with weight loss affect dyslipidemia in obese patients Citation[3].
Tumor necrosis factor-α
TNF-α may be increased with excessive body fat, and may promote an increase in FFAs, resulting in lipotoxicity and glucose intolerance in peripheral tissues, as well as a decrease in pancreatic insulin secretion Citation[205]. TNF-α’s interaction with other adipocyte factors, such as leptin and adiponectin, may also contribute to Type 2 DM Citation[48]. Weight loss through a hypocaloric diet and increased physical exercise in obese patients may cause TNF-α levels to trend downward Citation[143].
Visceral adipose tissue-derived serine protease inhibitor
Visceral adipose tissue-derived serine protease inhibitor is an adipokine predominantly secreted by visceral fat that increases insulin sensitivity Citation[206,207], normalizes glucose levels, and reverses altered expression of genes relevant to insulin resistance such as leptin, resistin, TNF-α, glucose transporter-4, and adiponectin Citation[208]. The effects of weight loss through a hypocaloric diet and increased physical exercise have not been well described.
Visfatin
Visfatin is preferentially produced by visceral fat, and may act as an insulin analog, and may play a role in the pathogenesis of Type 2 DM Citation[209]. Although the effects of weight loss through hypocaloric diets and increased physical exercise have not been well described, weight loss through gastric banding may decrease visfatin levels Citation[210].
Summary
In summary, in patients who respond to positive caloric balance with pathological increases or decreases in adipose factors (both inflammatory and noninflammatory) may develop adiposopathy, and thus promote metabolic abnormalities contributing to EFRMD. Weight loss and physical exercise can often reverse many of these pathological abnormalities, improve adipose tissue function, and thus serve as effective therapeutic modalities in treating EFRMD.
Expert commentary
Effective treatment of adiposopathy often associated with EFRMD can be achieved through increased recruitment of additional, functional adipose tissue, as might occur through the use of PPAR-γ agonists, which improve fat function through an increase in functional fat Citation[2,3,211]. Weight loss can also improve fat function through promoting the reduction in size of bloated, dysfunctional fat cells, as can be achieved through a hypocaloric diet, regular physical exercise, as well as anti-obesity agents that have been reviewed previously Citation[4,211,212], some of which may improve EFRMD due to mechanisms beyond weight loss Citation[213,214]. Regardless of the approach, the goals towards treating patients with adiposopathy is to improve adipose tissue functional capacity through unfettered adipogenesis (if needed), improve adipose tissue-mediated nutrient metabolism, reduce systemic inflammation and correct the over or under production of clinically relevant adipocyte factors which may all contribute to EFRMD, such as Type 2 DM, hypertension, dyslipidemia and even CHD Citation[3].
Five-year view
Among the areas most in need of future research is the elucidation of the manner in which adipose tissue interacts or undergoes ‘cross-talk’ with other major organ systems, including the CNS, peripheral nervous system, immune system, skeletal muscle, cardiovascular system, liver, gastrointestinal system (including pancreas), adrenal cortex, thyroid and other tissues.
For example, with regard to the nervous system, body fat function can affect the brain and peripheral nervous system function (as previously described with leptin signaling, as just one example) Citation[215]. In turn, the brain function can also affect fat function, in that both efferent and afferent innervation of adipose tissue can affect processes involved with fat regulation Citation[216]. With regard to skeletal muscle, it has been shown that the insulin resistance found in some patients may be predominantly due to impaired fat oxidation as opposed to increased FFA muscle uptake resulting from adiposopathy Citation[217,218]. In other words, during positive caloric balance, if adiposopathy ensues with increased FFA release, skeletal muscle may not always be flexible in its ability to modulate lipid oxidation, as the result of genetic limitations or impairment imposed by adiposopathy or other causes. In this state of ‘metabolic inflexibility’, intramyocyte triglycerides are increased, contributing to muscle insulin resistance Citation[219]. Thus, for many patients, it is likely that EFRMD may occur through both adipose tissue and peripheral tissue limitations and/or dysfunctions. This potential for a pathological partnership is supported by the finding that adipose tissue and muscle appear to have communications that might serve to worsen (or alternatively improve) EFRMD Citation[220]. In fact, it is probable that it is the simultaneous dysfunction of one or more organ systems that best accounts for the clinical expression and severity of EFRMD that is initiated by the adiposopathic process, and potentially influenced by interorgan communication. However, the exact nature of most body organ ‘cross-talk’ has yet to be comprehensively defined, and the precise relationship between multiple organ systems and the development of EFRMD, as well as how various adiposopathy interventions may improve such communications, is a priority for future research.
Table 1. Select comparisons of subcutaneous, peripheral fat versus visceral fat functions* Citation[18,19,223–230].
Table 2. Adiposopathy interventions and their effects upon select adipocyte factors that may contribute to diabetes mellitus Citation[3].
Table 3. Adiposopathy interventions and their effects upon select adipocyte factors that may contribute to hypertension Citation[3].
Table 4. Adiposopathy interventions and their effects upon select adipocyte factors that may contribute to dyslipidemia Citation[3].
Box 1. Examples of adipocyte factors relevant to adipogenesis.
Transcription factors:
| • | Adipocyte determination and differentiation factor-1/SREBP-1, CCAAT/enhancer binding proteins, cyclin D, E2F proteins and peroxisome proliferator-activated receptor γ and coactivator-1 | ||||
Extracellular matrix/cytoskeleton factors:
| • | Actin, collagen-binding protein, collagens, fibroblast growth factor, fibronectin, gelsolin, laminin, matrix metalloproteases, myocin, nidogen, osteonectin and tubulin | ||||
Adipose tissue proadipogenic factors:*
| • | Acylation stimulating protein, adipocyte plasma membrane associated protein, adipogenin, adiponectin, agouti protein, angiotensin II and angiotensinogen (promotes differentiation), angiotensin-converting enzyme, autotaxin (promotes proliferation), bone morphogenic protein, cathepsin S, cyclic adenosine monophosphate, epidermal growth factor (promotes adipogenesis in differentiated adipocytes), estrogens, free fatty acids, galectin 12 (promotes differentiation), heptatic growth factor (?), insulin-like growth factor, leptin‡, leukemia inhibitory factor‡, lipin, lysophosphosphatidic acid (promotes differentiation), macrophage colony stimulating factor, neuronatin, nitric oxide, phosphoinositide 3-kinase, plasminogen activator inhibitor-1‡, prolactin, prostaglandin E2, prostaglandin I2, resistin‡ and retinoids‡ | ||||
Adipose tissue antiadipogenic factors:*
| • | Androgens, angiotensin II and angiotensinogen (inhibits preadipocyte recruitment), angiotensinogen (inhibits preadipocyte recruitment), autotaxin (inhibits differentiation), ceramide, cystatin c, epidermal growth factor (inhibits preadipocyte differentiation into mature adipocytes), galectin 12 (inhibits proliferation), insulin-like growth factor binding protein, interleukins-1, -6, -11, leptin‡, leukemia inhibitory factor‡, lysophophosphatidic acid (inhibits differentiation), necdin, plasminogen activator inhibitor-1‡, preadipocyte factor-1, prostaglandin F2, protein kinase–mitogen-activated protein kinase, resistin‡, resistin-like molecules, retinoids‡, transforming growth factor-β, tumor necrosis factor-α | ||||
Box 2. Example of adipocyte factors that may be related to excessive fat-related metabolic disease.
Glucose metabolism
| • | 3 β-hydroxysteroid dehydrogenase, 11 β-hydroxysteroid dehydrogenase type 1, 17 α-hydroxylase, acylation-stimulating protein, adenosine, adiponectin, adipsin, angiotensin I and II, angiotensin-converting enzyme, angiotensinogen, apolipoprotein C1, apolipoprotein D, autotaxin, ceramide, clatherin, complement factors C3 and B, C-reactive protein, dynamin, ectonucleotide pyrophosphatase/phosphodiesterase-1, endothelin, flotillin, free fatty acids, glucose transporter 4, glutamine, glycerol, hormone-sensitive lipase, hypoxia inducible factor-1, insulin-like growth factor-1, insulin-like growth factor binding protein, interleukins, leptin, lipin, lysophospholipids, neuronatin, nitric oxide synthase, omentin, perilipin, phosphoenolpyrovate carboxykinase, phosphoinositide 3 kinase, plasminogen activator inhibitor-1, protein kinase–mitogen-actived protein kinase, protein kinase B, protein kinase C, resistin, resistin-like molecules, retinol-binding protein, tumor necrosis factor-α, visceral adipose tissue-derived serpin and visfatin | ||||
Vascular tone and blood pressure
| • | 11 β-hydroxysteroid dehydrogenase type 1, 3, β-hydroxysteroid dehydrogenase, adiponectin, adrenomedullin, angiotensin I and II, angiotensin-converting enzyme, angiotensinogen, apelin, cathepsins D and G, chymase, endothelin, free fatty acids, interleukin-6, leptin, nitric oxide synthase, prostaglandins, renin and tonin | ||||
Lipid metabolism
| • | 11 β-hydroxysteroid dehydrogenase type 1, acetyl-coenzyme A carboxylase, acylation-stimulating protein, adenosine, adiponectin, adiponutrin, adipophilin, adipsin, agouti protein, androgens, angiotensin I and II, angiotensinogen, apolipoproteins C1, D and E, aquaporin 7, calveolin-1, cholesteryl ester-transfer protein, desnutrin, estrogens, fasting-induced adipose factor, fatty acid binding protein, fatty acid synthase, fatty acid translocase, fatty acid transport protein, free fatty acids, glycerol, hormone-sensitive lipase, interleukin 11, leptin, lipin, lipoprotein lipase, perilipin, phosphoenolpyruvate carboxykinase, phospholipid transfer protein, prostaglandin E2, protein kinase A, S3-12, sex hormones and associated enzymes, stearoyl-CoA desaturase, tail interacting protein 47 and tumor necrosis factor-α. | ||||
Immunity/inflammation
| • | Acylation stimulating protein, adiponectin, adipsin, α-1 acid glycoprotein, annexin-1, bone morphogenic factor, ceruloplasmin, complement factors C3 and B, C-Reactive protein, eicosanoids (prostaglandins), eotaxin, haptoglobin, interferon inducible protein, interleukins, interleukin-receptor antagonist, leptin, macrophage colony stimulating factor, macrophage migration inhibitory factor, metallothionein, monocyte chemoattractant protein-1, neutrophil gelatinase-associated lipocalin, nitric oxide, normal T-cell expressed and secreted, pentraxin-3, plasminogen activator inhibitor-1, regulated on activation, resistin, serum amyloid A, stromal derived factor-1, transforming growth factor-β, tumor necrosis factor-α, vascular adhesion protein-1 and vascular cell adhesion molecule-1 | ||||
Steroid hormone factors
| • | 3 α-hyroxysteroid dehydrogenase, 3 β-hydroxysteroid dehydrogenase, 5 α-reductase, 11 β-hydroxysteoid dehydrogenase, 17 α-hydroxylase, 17 β-hydroxysteroid dehydrogenase, androgens, cytochrome p450-dependent aromatase, estrogens and urindine diphosphate glucuronosyltransferase | ||||
Angiogenesis
| • | Autotaxin, hypoxia inducible factor-1, leptin, monobutyrin, nitric acid synthase, nitric oxide synthase, osteonectin, pigment epithelium-derived factor, plasminogen activator inhibitor-1, prostaglandin I2, tissue factor and vascular endothelial growth factor | ||||
Coagulation/fibrinolysis
| • | Annexin 2 and 5, plasminogen activator inhibitor-1 and tissue factor | ||||
Key issues
| • | During positive caloric balance, unimpaired recruitment, proliferation and differentiation of new, ‘healthy’ adipocytes (such as in subcutaneous, peripheral fat) may maintain normal metabolic processes, preventing onset of excessive fat-related metabolic diseases (EFRMD), despite fat weight gain. The recruitment, proliferation and differentiation of adipocytes leading to fat accumulation in visceral fat may lead to EFRMD. Limitations or impaired recruitment and proliferation of new, ‘healthy’ adipocytes may lead to excessive adipocyte hypertrophy and adiposopathy of various fat depots and thus promote EFRMD. | ||||
| • | Improvements in EFRMD can be achieved through a reduction in ‘sick fat’ by hypocaloric diets in overweight or obese patients, which downregulates adipogenesis as a natural physiological response. Alternatively, improvements in EFRMD can be achieved by an increase in ‘healthy fat’, by peroxisome proliferator-activated receptor (PPAR)-γ agonists, which upregulates adipogenesis. | ||||
| • | Subcutaneous, peripheral and visceral fat differ with regard to metabolic activity and metabolic functions. | ||||
| • | The dysfunction of subcutaneous, peripheral and visceral fat, as well as the dysfunction of other fat depots, such as abdominal subcutaneous, intraorgan and periorgan fat, all contribute to EFRMD (including atherosclerotic cardiovascular disease). | ||||
| • | A reduction in visceral fat through weight loss, diet, physical exercise, PPAR-γ agents, orlistat, sibutramine and cannabinoid (CB)-1 receptor antagonists and CB-receptor antagonists is associated with improvements in various metabolic parameters associated with EFRMD. | ||||
| • | Increased net release of free fatty acids (FFAs) is lipotoxic to major body organs, such as muscle, liver and pancreas, and thus helps promote dysmetabolisms contributing to EFRMD. | ||||
| • | Starvation or abrupt weight loss increases adipocyte triglyceride lipolysis, with an increased energy supply of FFAs to skeletal muscle. The glycerol formed by triglyceride lipolysis is utilized by the liver for gluconeogensis Citation[221], which is a process stimulated by increased circulating FFA, as is found with starvation. | ||||
| • | Gradual weight loss through diet and physical exercise reduces FFA levels, reduces lipotoxicity and thus improves metabolic abnormalities promoting EFRMD. | ||||
| • | PPAR-γ agonists, orlistat, possibly sibutramine and CB-1 receptor antagonists also reduce FFA levels, which helps account for improvements in metabolic parameters found with such therapies. | ||||
| • | Adipocyte hypertrophy and excessive adipose tissue growth may result in pathological increases and decreases in adipocyte factors. | ||||
| • | Pathological increases, decreases or pathological limitations upon the production and activity of adipocyte factors may promote adiposopathy. | ||||
| • | Interventions that correct the pathological activity of adipocyte factors may prevent or improve EFRMD. | ||||
| • | Inflammation resulting from adiposopathy contributes to EFRMD. | ||||
| • | During positive caloric balance, visceral fat accumulation and hypertrophy and subcutaneous, peripheral fat hypertrophy may increase the release of proinflammatory adipocyte factors such as adipocytokines, acute-phase response factors, chemoattractants for inflammatory cells, complement proteins and eicosanoids while the secretion of anti-inflammatory adipocyte factors (such as adiponectin) may be decreased. | ||||
| • | Interventions that improve fat function may reduce the proinflammatory adipose tissue response and increase an anti-inflammatory response, which helps account for the improvement in EFRMD associated with improved fat function. | ||||
| • | Adipose tissue interacts or undergoes ‘cross-talk’ with other major organ systems, including the CNS, peripheral nervous system, immune system, skeletal muscle, cardiovascular system, liver, gastrointestinal system (including pancreas), adrenal cortex, thyroid and other tissues. | ||||
| • | For many, if not most, patients, while adiposopathy may be the initial pathophysiological promoter of systemic dysmetabolisms, it is likely that EFRMD is ultimately the result of a pathological partnership involving adipose tissue limitations and/or dysfunctions, coupled with limitations and/or dysfunctions of other organ systems, including impaired communication or ‘cross-talk’. | ||||
| • | Much research needs to be conducted in order to better understand how adiposopathy interventions may improve the interactions of adipose tissue with other organ systems, and thus improve EFRMD. | ||||
Notes
*Does not necessarily include adipocyte factors already listed in ‘Transcription factors’ and ‘Extracellular matrix/cytoskeleton factors’ listed in this box, or the “Angiogenesis factors” listed in Box 2, many of which also help regulate adipogenesis. Also, individual adipocyte factors may promote or inhibit adipocyte proliferation or differentiation or both, or have differing effects upon these two processes, which are integral to adipogenesis.
‡As with other data concerning resistin and metabolic disease, conflicting reports suggest that resistin may enhance or impair adipogenesis. Leukemia inhibitory factor, plasminogen activator inhibitor-1, retinoids also have unclear, and sometimes conflicting data regarding their effects upon adipogenesis. Even the effects of leptin upon adipogenesis are not always consistent in the medical literature
Adipose tissue has many of the same mechanical factors necessary for cellular and tissue functions, and their appropriate activity is no less important to metabolic health than that of other body organs. The normal production, activity, and/or secretion of adipocyte factors are important for normal adipose tissue function. The abnormal production, activity, and/or secretion of adipocyte factors results in dysfunctional fat function – adiposopathy. For example, hypertrophy of adipocytes impairs normal function, and if adipose tissue exceeds its vascular supply, then stimuli such as hypoxia may promote an inflammatory response.
Related Research Data
References
- Bays H, Abate N, Chandalia M. Adiposopathy: Sick fat causes high blood sugar, high blood pressure, and dyslipidemia. Future Cardiol.1(1), 39–59 (2005).
- Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin.Endocrinol.Metab.89(2), 463–478 (2004).
- Bays H, Ballantyne C. Adiposopathy: why do adiposity and obesity cause metabolic disease? Future Lipidol.1(4), 389–420 (2006).
- Bays HE. Current and investigational antiobesity agents and obesity therapeutic treatment targets. Obes. Res.12(8), 1197–1211 (2004).
- Bays H, Dujovne CA. Adiposopathy is a more rational treatment target for metabolic disease than obesity alone. Curr. Atheroscler. Rep.8(2), 144–156 (2006).
- Vidal-Puig AJ, Considine RV, Jimenez-Linan M et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Invest99(10), 2416–2422 (1997).
- Viguerie N, Vidal H, Arner P et al. Adipose tissue gene expression in obese subjects during low-fat and high-fat hypocaloric diets. Diabetologia48(1), 123–131 (2005).
- Gurnell M. Peroxisome proliferator-activated receptor γ and the regulation of adipocyte function: lessons from human genetic studies. Best Pract. Res. Clin. Endocrinol. Metab.19(4), 501–523 (2005).
- Miyazaki Y, Mahankali A, Matsuda M et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J. Clin.Endocrinol.Metab.87(6), 2784–2791 (2002).
- Jbilo O, Ravinet-Trillou C, Arnone M et al. The CB1 receptor antagonist rimonabant reverses the diet-induced obesity phenotype through the regulation of lipolysis and energy balance. FASEB J.19(11), 1567–1569 (2005).
- Gary-Bobo M, Elachouri G, Scatton B, Le Fur G, Oury-Donat F, Bensaid M. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits cell proliferation and increases markers of adipocyte maturation in cultured mouse 3T3 F442A preadipocytes. Mol. Pharmacol.69(2), 471–478 (2006).
- Osei-Hyiaman D, Harvey-White J, Batkai S, Kunos G. The role of the endocannabinoid system in the control of energy homeostasis. Int. J. Obes. (Lond) 30(Suppl. 1), S33–S38 (2006).
- Pagotto U, Vicennati V, Pasquali R. The endocannabinoid system and the treatment of obesity. Ann. Med.37(4), 270–275 (2005).
- Serazin V, Dieudonne MN, Morot M, de Mazancourt P, Giudicelli Y. cAMP-positive regulation of angiotensinogen gene expression and protein secretion in rat adipose tissue. Am. J. Physiol. Endocrinol. Metab.286(3), E434–E438 (2004).
- Abate N, Garg A. Heterogeneity in adipose tissue metabolism: causes, implications and management of regional adiposity. Prog. Lipid Res.34(1), 53–70 (1995).
- Arner P. Regional differences in protein production by human adipose tissue. Biochem. Soc. Trans.29(Pt 2), 72–75 (2001).
- Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev.23(2), 201–229 (2002).
- Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr. Rev.21(6), 697–738 (2000).
- Dusserre E, Moulin P, Vidal H. Differences in mRNA expression of the proteins secreted by the adipocytes in human subcutaneous and visceral adipose tissues. Biochim. Biophys. Acta1500(1), 88–96 (2000).
- McCarty MF. Modulation of adipocyte lipoprotein lipase expression as a strategy for preventing or treating visceral obesity. Med. Hypotheses57(2), 192–200 (2001).
- Matsuzawa Y. Therapy Insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nat. Clin. Pract.3(1), 35–42 (2006).
- Liese AD, Mayer-Davis EJ, Tyroler HA et al. Development of the multiple metabolic syndrome in the ARIC cohort: joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol.7(6), 407–416 (1997).
- Janssen I, Katzmarzyk PT, Ross R. Waist circumference and not body mass index explains obesity-related health risk. Am. J. Clin. Nutr.79(3), 379–384 (2004).
- Zhu S, Heymsfield SB, Toyoshima H, Wang Z, Pietrobelli A, Heshka S. Race-ethnicity-specific waist circumference cutoffs for identifying cardiovascular disease risk factors. Am. J. Clin. Nutr.81(2), 409–415 (2005).
- Baker AR, Silva NF, Quinn DW et al. Human epicardial adipose tissue expresses a pathogenic profile of adipocytokines in patients with cardiovascular disease. Cardiovasc. Diabetol.5, 1 (2006).
- Zamboni M, Armellini F, Turcato E et al. Effect of weight loss on regional body fat distribution in premenopausal women. Am. J. Clin. Nutr.58(1), 29–34 (1993).
- Shadid S, Jensen MD. Effects of pioglitazone versus diet and exercise on metabolic health and fat distribution in upper body obesity. Diabetes Care26(11), 3148–3152 (2003).
- Kay SJ, Fiatarone Singh MA. The influence of physical activity on abdominal fat: a systematic review of the literature. Obes. Rev.7(2), 183–200 (2006).
- Kopelman PG. The effects of weight loss treatments on upper and lower body fat. Int. J. Obes. Relat. Metab. Disord.21(8), 619–625 (1997).
- O’Leary VB, Marchetti CM, Krishnan RK, Stetzer BP, Gonzalez F, Kirwan JP. Exercise-induced reversal of insulin resistance in obese elderly is associated with reduced visceral fat. J. Appl. Physiol.100(5), 1584–1589 (2006).
- Ross R, Janssen I, Dawson J et al. Exercise-induced reduction in obesity and insulin resistance in women: a randomized controlled trial. Obes. Res.12(5), 789–798 (2004).
- Larson-Meyer DE, Heilbronn LK, Redman LM et al. Effect of calorie restriction with or without exercise on insulin sensitivity, β-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes Care29(6), 1337–1344 (2006).
- Miyashita Y, Koide N, Ohtsuka M et al. Beneficial effect of low carbohydrate in low calorie diets on visceral fat reduction in type 2 diabetic patients with obesity. Diabetes Res. Clin. Pract.65(3), 235–241 (2004).
- Tiikkainen M, Bergholm R, Rissanen A et al. Effects of equal weight loss with orlistat and placebo on body fat and serum fatty acid composition and insulin resistance in obese women. Am. J. Clin. Nutr.79(1), 22–30 (2004).
- Kelley DE, Kuller LH, McKolanis TM, Harper P, Mancino J, Kalhan S. Effects of moderate weight loss and orlistat on insulin resistance, regional adiposity, and fatty acids in type 2 diabetes. Diabetes Care27(1), 33–40 (2004).
- Van Gaal LF, Wauters MA, Peiffer FW, De Leeuw I. Sibutramine and fat distribution: is there a role for pharmacotherapy in abdominal/visceral fat reduction? Int. J. Obes. Relat. Metab. Disord.22(Suppl. 1), S38–S40 (1998).
- Despres JP. Intra-abdominal obesity: an untreated risk factor for Type 2 diabetes and cardiovascular disease. J. Endocrinol. Invest.29(3 Suppl.), 77–82 (2006).
- Roche R, Hoareau L, Bes-Houtmann S et al. Presence of the cannabinoid receptors, CB1 and CB2, in human omental and subcutaneous adipocytes. Histochem. Cell Biol.126(2), 177–187 (2006).
- Grundy SM. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab.89(6), 2595–2600 (2004).
- Wood IS, Trayhurn P. Adipokines and the signaling role of adipose tissue in inflammation and obesity. Future Lipidol.1(1), 81–89 (2006).
- Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int. J. Obes. Relat. Metab. Disord.28(Suppl. 4) S12–S21 (2004).
- Fagot-Campagna A, Balkau B, Simon D et al. High free fatty acid concentration: an independent risk factor for hypertension in the Paris Prospective Study. Int. J. Epidemiol.27(5), 808–813 (1998).
- de Jongh RT, Serne EH, Ijzerman RG, de Vries G, Stehouwer CD. Free fatty acid levels modulate microvascular function: relevance for obesity-associated insulin resistance, hypertension, and microangiopathy. Diabetes53(11), 2873–2882 (2004).
- Egan BM. Insulin resistance and the sympathetic nervous system. Curr. Hypertens. Rep.5(3), 247–254 (2003).
- Bays HE, McGovern ME. Once-daily niacin extended release/lovastatin combination tablet has more favorable effects on lipoprotein particle size and subclass distribution than atorvastatin and simvastatin. Prev. Cardiol.6(4), 179–188 (2003).
- Yu YH, Ginsberg HN. Adipocyte signaling and lipid homeostasis: sequelae of insulin-resistant adipose tissue. Circ. Res.96(10), 1042–1052 (2005).
- Bryson JM, King SE, Burns CM, Baur LA, Swaraj S, Caterson ID. Changes in glucose and lipid metabolism following weight loss produced by a very low calorie diet in obese subjects. Int. J. Obes. Relat. Metab. Disord.20(4), 338–345 (1996).
- Faraj M, Lu HL, Cianflone K. Diabetes, lipids, and adipocyte secretagogues. Biochem. Cell Biol.82(1), 170–190 (2004).
- Seshadri P, Samaha FF, Stern L, Chicano KL, Daily DA, Iqbal N. Free fatty acids, insulin resistance, and corrected qt intervals in morbid obesity: effect of weight loss during 6 months with differing dietary interventions. Endocr. Pract.11(4), 234–239 (2005).
- Corbi GM, Carbone S, Ziccardi P et al. FFAs and QT intervals in obese women with visceral adiposity: effects of sustained weight loss over 1 year. J. Clin. Endocrinol. Metab.87(5), 2080–2083 (2002).
- De Glisezinski I, Moro C, Pillard F et al. Aerobic training improves exercise-induced lipolysis in SCAT and lipid utilization in overweight men. Am. J. Physiol. Endocrinol. Metab.285(5), E984–E990 (2003).
- Esposito K, Pontillo A, Di Palo C et al. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA289(14), 1799–1804 (2003).
- Marniemi J, Vuori I, Kinnunen V, Rahkila P, Vainikka M, Peltonen P. Metabolic changes induced by combined prolonged exercise and low-calorie intake in man. Eur. J. Appl. Physiol. Occup. Physiol.53(2), 121–127 (1984).
- Noakes M, Keogh JB, Foster PR, Clifton PM. Effect of an energy-restricted, high-protein, low-fat diet relative to a conventional high-carbohydrate, low-fat diet on weight loss, body composition, nutritional status, and markers of cardiovascular health in obese women. Am. J. Clin. Nutr.81(6), 1298–1306 (2005).
- Sasaki J, Shindo M, Tanaka H, Ando M, Arakawa K. A long-term aerobic exercise program decreases the obesity index and increases the high density lipoprotein cholesterol concentration in obese children. Int. J. Obes.11(4), 339–345 (1987).
- Williams PT, Krauss RM, Vranizan KM, Albers JJ, Wood PD. Effects of weight-loss by exercise and by diet on apolipoproteins A-I and A-II and the particle-size distribution of high-density lipoproteins in men. Metabolism41(4), 441–449 (1992).
- Slyper AH. Low-density lipoprotein density and atherosclerosis. Unraveling the connection. JAMA272(4), 305–308 (1994).
- DeFronzo RA. Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int. J. Clin. Pract.143(Suppl.), 9–21 (2004).
- Boden G, Laakso M. Lipids and glucose in type 2 diabetes: what is the cause and effect? Diabetes Care27(9), 2253–2259 (2004).
- Kiortsis DN, Filippatos TD, Elisaf MS. The effects of orlistat on metabolic parameters and other cardiovascular risk factors. Diabetes Metab.31(1), 15–22 (2005).
- Day C, Bailey CJ. Effect of the antiobesity agent sibutramine in obese-diabetic ob/ob mice. Int. J. Obes. Relat. Metab. Disord.22(7), 619–623 (1998).
- McLaughlin T, Abbasi F, Lamendola C, Kim HS, Reaven GM. Metabolic changes following sibutramine-assisted weight loss in obese individuals: role of plasma free fatty acids in the insulin resistance of obesity. Metabolism50(7), 819–824 (2001).
- Ravinet Trillou C, Arnone M, Delgorge C et al. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am. J. Physiol. Regul. Integr. Comp. Physiol.284(2), R345–R353 (2003).
- Hissin PJ, Foley JE, Wardzala LJ et al. Mechanism of insulin-resistant glucose transport activity in the enlarged adipose cell of the aged, obese rat. J. Clin. Invest.70(4), 780–790 (1982).
- Norris SL, Zhang X, Avenell A et al. Long-term effectiveness of lifestyle and behavioral weight loss interventions in adults with type 2 diabetes: a meta-analysis. Am. J. Med.117(10), 762–774 (2004).
- Aucott L, Poobalan A, Smith WC et al. Weight loss in obese diabetic and non-diabetic individuals and long-term diabetes outcomes--a systematic review. Diabetes Obes. Metab.6(2), 85–94 (2004).
- Boden G, Sargrad K, Homko C, Mozzoli M, Stein TP. Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Ann. Intern. Med.142(6), 403–411 (2005).
- Bruun JM, Helge JW, Richelsen B, Stallknecht B. Diet and Exercise Reduce Low-grade Inflammation and Macrophage Infiltration in Adipose Tissue but not in Skeletal Muscle in Severely Obese Subjects. Am. J. Physiol. Endocrinol. Metab.290(5), E961–E967 (2005).
- Straznicky NE, Louis WJ, McGrade P, Howes LG. The effects of dietary lipid modification on blood pressure, cardiovascular reactivity and sympathetic activity in man. J. Hypertens.11(4), 427–437 (1993).
- Aucott L, Poobalan A, Smith WC, Avenell A, Jung R, Broom J. Effects of weight loss in overweight/obese individuals and long-term hypertension outcomes: a systematic review. Hypertension45(6), 1035–1041 (2005).
- Ruano M, Silvestre V, Castro R et al. Morbid obesity, hypertensive disease and the renin-angiotensin-aldosterone axis. Obes. Surg.15(5), 670–676 (2005).
- Stone NJ, Kushner R. Effects of dietary modification and treatment of obesity. Emphasis on improving vascular outcomes. Med. Clin. North Am.84(1), 95–122 (2000).
- Chen AK, Roberts CK, Barnard RJ. Effect of a short-term diet and exercise intervention on metabolic syndrome in overweight children. Metabolism55(7), 871–878 (2006).
- Dahlman I, Linder K, Arvidsson Nordstrom E et al. Changes in adipose tissue gene expression with energy-restricted diets in obese women. Am. J. Clin. Nutr.81(6), 1275–1285 (2005).
- Kannisto K, Pietilainen KH, Ehrenborg E et al. Overexpression of 11β-hydroxysteroid dehydrogenase-1 in adipose tissue is associated with acquired obesity and features of insulin resistance: studies in young adult monozygotic twins. J. Clin. Endocrinol. Metab.89(9), 4414–4421 (2004).
- Tomlinson JW, Moore JS, Clark PM, Holder G, Shakespeare L, Stewart PM. Weight loss increases 11β-hydroxysteroid dehydrogenase type 1 expression in human adipose tissue. J. Clin. Endocrinol. Metab.89(6), 2711–2716 (2004).
- Valsamakis G, Anwar A, Tomlinson JW et al. 11β-hydroxysteroid dehydrogenase type 1 activity in lean and obese males with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab.89(9), 4755–4761 (2004).
- Putignano P, Pecori Giraldi F, Cavagnini F. Tissue-specific dysregulation of 11β-hydroxysteroid dehydrogenase type 1 and pathogenesis of the metabolic syndrome. J. Endocrinol. Invest.27(10), 969–974 (2004).
- Engeli S, Bohnke J, Feldpausch M et al. Regulation of 11β-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes. Res.12(1), 9–17 (2004).
- Coutinho AE, Campbell JE, Fediuc S, Riddell MC. Effect of voluntary exercise on peripheral tissue glucocorticoid receptor content and the expression and activity of 11{β}-HSD1 in the Syrian hamster. J. Appl. Physiol.100(5), 1483–1488 (2005).
- Wake DJ, Walker BR. 11 β-hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Mol. Cell Endocrinol.215(1–2), 45–54 (2004).
- Masuzaki H, Yamamoto H, Kenyon CJ et al. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J. Clin. Invest.112(1), 83–90 (2003).
- Morton NM, Paterson JM, Masuzaki H et al. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 β-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes53(4), 931–938 (2004).
- Walker BR. 11β-hydroxysteroid dehydrogenase type 1 in obesity. Obes. Res.12(1), 1–3 (2004).
- Masuzaki H, Flier JS. Tissue-specific glucocorticoid reactivating enzyme, 11 β-hydroxysteroid dehydrogenase type 1 (11 β-HSD1) – a promising drug target for the treatment of metabolic syndrome. Curr. Drug Targets Immune Endocr. Metabol. Disord.3(4), 255–262 (2003).
- Cianflone K, Xia Z, Chen LY. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim. Biophys. Acta1609(2), 127–143 (2003).
- Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab.89(6), 2548–2556 (2004).
- Ahren B, Havel PJ, Pacini G, Cianflone K. Acylation stimulating protein stimulates insulin secretion. Int. J. Obes. Relat. Metab. Disord.27(9), 1037–1043 (2003).
- Havel PJ. Update on adipocyte hormones: regulation of energy balance and carbohydrate/lipid metabolism. Diabetes53(Suppl. 1) S143–S151 (2004).
- Faraj M, Sniderman AD, Cianflone K. ASP enhances in situ lipoprotein lipase activity by increasing fatty acid trapping in adipocytes. J. Lipid Res.45(4), 657–666 (2004).
- Maslowska M, Wang HW, Cianflone K. Novel roles for acylation stimulating protein/C3adesArg: a review of recent in vitro and in vivo evidence. Vitam. Horm.70, 309–332 (2005).
- Faraj M, Havel PJ, Phelis S, Blank D, Sniderman AD, Cianflone K. Plasma acylation-stimulating protein, adiponectin, leptin, and ghrelin before and after weight loss induced by gastric bypass surgery in morbidly obese subjects. J. Clin. Endocrinol. Metab.88(4), 1594–1602 (2003).
- Sniderman AD, Cianflone KM, Eckel RH. Levels of acylation stimulating protein in obese women before and after moderate weight loss. Int. J. Obes.15(5), 333–336 (1991).
- Schrauwen P, Hesselink MK, Jain M, Cianflone K. Acylation-stimulating protein: effect of acute exercise and endurance training. Int. J. Obes.(Lond.)29(6), 632–638 (2005).
- Pittas AG, Joseph NA, Greenberg AS. Adipocytokines and insulin resistance. J. Clin. Endocrinol. Metab.89(2), 447–452 (2004).
- Kopp HP, Krzyzanowska K, Mohlig M, Spranger J, Pfeiffer AF, Schernthaner G. Effects of marked weight loss on plasma levels of adiponectin, markers of chronic subclinical inflammation and insulin resistance in morbidly obese women. Int. J. Obes.(Lond.)29(7), 766–771 (2005).
- Valsamakis G, McTernan PG, Chetty R et al. Modest weight loss and reduction in waist circumference after medical treatment are associated with favorable changes in serum adipocytokines. Metabolism53(4), 430–434 (2004).
- Balagopal P, George D, Yarandi H, Funanage V, Bayne E. Reversal of obesity-related hypoadiponectinemia by lifestyle intervention: a controlled, randomized study in obese adolescents. J. Clin. Endocrinol. Metab.90(11), 6192–6197 (2005).
- Merl V, Peters A, Oltmanns KM et al. Serum adiponectin concentrations during a 72-hour fast in over- and normal-weight humans. Int. J. Obes.(Lond.)29(8), 998–1001 (2005).
- Anderlova K, Kremen J, Dolezalova R et al. The influence of very-low-calorie-diet on serum leptin, soluble leptin receptor, adiponectin and resistin levels in obese women. Physiol. Res.55(3), 277–283(2005).
- Mousavinasab F, Tahtinen T, Jokelainen J et al. Lack of increase of serum adiponectin concentrations with a moderate weight loss during six months on a high-caloric diet in military service among a young male Finnish population. Endocrine26(1), 65–69 (2005).
- Xydakis AM, Case CC, Jones PH et al. Adiponectin, inflammation, and the expression of the metabolic syndrome in obese individuals: the impact of rapid weight loss through caloric restriction. J. Clin. Endocrinol. Metab.89(6), 2697–2703 (2004).
- Nassis GP, Papantakou K, Skenderi K et al. Aerobic exercise training improves insulin sensitivity without changes in body weight, body fat, adiponectin, and inflammatory markers in overweight and obese girls. Metabolism54(11), 1472–1479 (2005).
- Huang KC, Chen CL, Chuang LM, Ho SR, Tai TY, Yang WS. Plasma adiponectin levels and blood pressures in nondiabetic adolescent females. J. Clin. Endocrinol. Metab.88(9), 4130–4134 (2003).
- Coatmellec-Taglioni G, Ribiere C. Factors that influence the risk of hypertension in obese individuals. Curr. Opin. Nephrol. Hypertens.12(3), 305–308 (2003).
- Rochlitz H, Akpulat S, Bobbert T et al. [Significance of biomarkers for metabolic syndrome during weight reduction]. Dtsch. Med. Wochenschr.130(17), 1061–1066 (2005).
- Matsubara M, Maruoka S, Katayose S. Decreased plasma adiponectin concentrations in women with dyslipidemia. J. Clin. Endocrinol. Metab.87(6), 2764–2769 (2002).
- Maslowska M, Vu H, Phelis S, Sniderman AD, Rhode BM, Blank D, Cianflone K. Plasma acylation stimulating protein, adipsin and lipids in non-obese and obese populations. Eur. J. Clin. Invest.29(8), 679–686 (1999).
- Miner JL. The adipocyte as an endocrine cell. J. Anim. Sci.82(3), 935–941 (2004).
- Pomeroy C, Mitchell J, Eckert E, Raymond N, Crosby R, Dalmasso AP. Effect of body weight and caloric restriction on serum complement proteins, including Factor D/adipsin: studies in anorexia nervosa and obesity. Clin. Exp. Immunol.108(3), 507–515 (1997).
- Mavri A, Stegnar M, Krebs M, Sentocnik JT, Geiger M, Binder BR. Impact of adipose tissue on plasma plasminogen activator inhibitor-1 in dieting obese women. Arterioscler. Thromb. Vasc. Biol.19(6), 1582–1587 (1999).
- Strazzullo P, Galletti F. Impact of the renin-angiotensin system on lipid and carbohydrate metabolism. Curr. Opin. Nephrol. Hypertens.13(3), 325–332 (2004).
- Engeli S, Bohnke J, Gorzelniak K et al. Weight loss and the renin-angiotensin-aldosterone system. Hypertension45(3), 356–362 (2005).
- Maeda S, Iemitsu M, Jesmin S, Miyauchi T. Acute exercise causes an enhancement of tissue renin-angiotensin system in the kidney in rats. Acta Physiol. Scand.185(1), 79–86 (2005).
- Rauramaa R, Kuhanen R, Lakka TA et al. Physical exercise and blood pressure with reference to the angiotensinogen M235T polymorphism. Physiol. Genomics10(2), 71–77 (2002).
- Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A et al. Human adipocytes secrete mineralocorticoid-releasing factors. Proc. Natl Acad. Sci. USA100(24), 14211–14216 (2003).
- Morse SA, Bravo PE, Morse MC, Reisin E. The heart in obesity-hypertension. Expert Rev. Cardiovasc. Ther.3(4), 647–658 (2005).
- Karlsson C, Lindell K, Ottosson M, Sjostrom L, Carlsson B, Carlsson LM. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II. J. Clin. Endocrinol. Metab.83(11), 3925–3929 (1998).
- Engeli S, Negrel R, Sharma AM. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension35(6), 1270–1277 (2000).
- Ferry G, Tellier E, Try A et al. Autotaxin is released from adipocytes, catalyzes lysophosphatidic acid synthesis, and activates preadipocyte proliferation. Up-regulated expression with adipocyte differentiation and obesity. J. Biol. Chem.278(20), 18162–18169 (2003).
- Boucher J, Quilliot D, Praderes JP et al. Potential involvement of adipocyte insulin resistance in obesity-associated up-regulation of adipocyte lysophospholipase D/autotaxin expression. Diabetologia48(3), 569–577 (2005).
- Summers SA, Nelson DH. A role for sphingolipids in producing the common features of type 2 diabetes, metabolic syndrome X, and Cushing's syndrome. Diabetes54(3), 591–602 (2005).
- Sawai H, Domae N, Okazaki T. Current status and perspectives in ceramide-targeting molecular medicine. Curr. Pharm. Des11(19), 2479–2487 (2005).
- Miura A, Kajita K, Ishizawa M et al Inhibitory effect of ceramide on insulin-induced protein kinase Czeta translocation in rat adipocytes. Metabolism52(1), 19–24 (2003).
- Long SD, Pekala PH. Lipid mediators of insulin resistance: ceramide signalling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytes. Biochem. J319(Pt 1) 179–184 (1996).
- Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J. Biol. Chem.280(20), 20148–20153 (2005).
- Adams JM, Pratipanawatr T, Berria R et al. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes53(1), 25–31 (2004).
- asimacopoulos-Jeannet F. Fat storage in pancreas and in insulin-sensitive tissues in pathogenesis of type 2 diabetes. Int. J. Obes. Relat. Metab. Disord.28(Suppl. 4) S53–S57 (2004).
- Helge JW, Dobrzyn A, Saltin B, Gorski J. Exercise and training effects on ceramide metabolism in human skeletal muscle. Exp. Physiol.89(1), 119–127 (2004).
- Gauthier B, Robb M, McPherson R. Cholesteryl ester transfer protein gene expression during differentiation of human preadipocytes to adipocytes in primary culture. Atherosclerosis142(2), 301–307 (1999).
- Bays H McKenney J Davidson M. Torcetrapib/Atorvastatin combination therapy. Expert Rev. Cardiovasc. Ther.3(5), 789–820 (2005).
- Ebenbichler CF, Laimer M, Kaser S et al. Relationship between cholesteryl ester transfer protein and atherogenic lipoprotein profile in morbidly obese women. Arterioscler. Thromb. Vasc. Biol.22(9), 1465–1469 (2002).
- Seip RL, Moulin P, Cocke T et al. Exercise training decreases plasma cholesteryl ester transfer protein. Arterioscler. Thromb.13(9), 1359–1367 (1993).
- Horowitz JF. Fatty acid mobilization from adipose tissue during exercise. Trends Endocrinol. Metab.14(8), 386–392 (2003).
- Large V, Reynisdottir S, Langin D et al. Decreased expression and function of adipocyte hormone-sensitive lipase in subcutaneous fat cells of obese subjects. J. Lipid Res.40(11), 2059–2066 (1999).
- Yeaman SJ. Hormone-sensitive lipase–new roles for an old enzyme. Biochem. J379(Pt 1), 11–22 (2004).
- Stich V, Harant I, De Glisezinski I et al. Adipose tissue lipolysis and hormone-sensitive lipase expression during very-low-calorie diet in obese female identical twins. J. Clin. Endocrinol. Metab.82(3), 739–744 (1997).
- Li Y, Bujo H, Takahashi K et al. Visceral fat: higher responsiveness of fat mass and gene expression to calorie restriction than subcutaneous fat. Exp. Biol. Med.228(10), 1118–1123 (2003).
- De Glisezinski I, Crampes F, Harant I et al. Endurance training changes in lipolytic responsiveness of obese adipose tissue. Am. J. Physiol.275(6 Pt 1), E951–E956 (1998).
- Lee DL, Sturgis LC, Labazi H et al. Angiotensin II Hypertension is Attenuated in Interleukin-6 Knockout Mice. Am. J. Physiol. Heart Circ. Physiol.290(3), H935–940 (2005).
- Orshal JM, Khalil RA. Interleukin-6 impairs endothelium-dependent NO-cGMP-mediated relaxation and enhances contraction in systemic vessels of pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol.286(6), R1013–R1023 (2004).
- Garanty-Bogacka B, Syrenicz M, Syrenicz A, Gebala A, Lulka D, Walczak M. Serum markers of inflammation and endothelial activation in children with obesity-related hypertension. Neuro. Endocrinol. Lett.26(3), 242–246 (2005).
- Monzillo LU, Hamdy O, Horton ES et al. Effect of lifestyle modification on adipokine levels in obese subjects with insulin resistance. Obes. Res.11(9), 1048–1054 (2003).
- Bertile F, Criscuolo F, Oudart H, Le Maho Y, Raclot T. Differences in the expression of lipolytic-related genes in rat white adipose tissues. Biochem. Biophys. Res. Commun.307(3), 540–546 (2003).
- Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes. Rev.2(4), 239–254 (2001).
- Siddals KW, Westwood M, Gibson JM, White A. IGF-binding protein-1 inhibits IGF effects on adipocyte function: implications for insulin-like actions at the adipocyte. J. Endocrinol.174(2), 289–297 (2002).
- Mauras N, Haymond MW. Are the metabolic effects of GH and IGF-I separable? Growth Horm. IGF Res.15(1), 19–27 (2005).
- Engstrom BE, Burman P, Holdstock C, Ohrvall M, Sundbom M, Karlsson FA. Effects of gastric bypass on the GH/IGF-I axis in severe obesity - and a comparison with GH deficiency. Eur. J. Endocrinol.154(1), 53–59 (2006).
- Butzow TL, Lehtovirta M, Siegberg R et al. The decrease in luteinizing hormone secretion in response to weight reduction is inversely related to the severity of insulin resistance in overweight women. J. Clin. Endocrinol. Metab.85(9), 3271–3275 (2000).
- Daly RM, Dunstan DW, Owen N, Jolley D, Shaw JE, Zimmet PZ. Does high-intensity resistance training maintain bone mass during moderate weight loss in older overweight adults with type 2 diabetes? Osteoporos. Int.16(12), 1703–1712 (2005).
- Havel PJ. Role of adipose tissue in body-weight regulation: mechanisms regulating leptin production and energy balance. Proc. Nutr. Soc.59(3), 359–371 (2000).
- Yildiz BO, Haznedaroglu IC. Rethinking leptin and insulin action: Therapeutic opportunities for diabetes. Int. J. Biochem. Cell Biol.38, 820–830 (2005).
- Persson-Sjogren S, Elmi A, Lindstrom P. Effects of leptin, acetylcholine and vasoactive intestinal polypeptide on insulin secretion in isolated ob/ob mouse pancreatic islets. Acta Diabetol.41(3), 104–112 (2004).
- Ceddia RB, Koistinen HA, Zierath JR, Sweeney G. Analysis of paradoxical observations on the association between leptin and insulin resistance. FASEB J.16(10), 1163–1176 (2002).
- Benomar Y, Rideau N, Crochet S, Derouet M, Taouis M. Leptin fully suppresses acetylcholine-induced insulin secretion and is reversed by tolbutamide in isolated perfused chicken pancreas. Horm. Metab. Res.35(2), 81–85 (2003).
- Zieba DA, Amstalden M, Maciel MN et al. Divergent effects of leptin on luteinizing hormone and insulin secretion are dose dependent. Exp. Biol. Med.228(3), 325–330 (2003).
- Frank LL, Sorensen BE, Yasui Y et al. Effects of exercise on metabolic risk variables in overweight postmenopausal women: a randomized clinical trial. Obes. Res.13(3), 615–625 (2005).
- Rosenbaum M, Goldsmith R, Bloomfield D et al. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J. Clin. Invest.115(12), 3579–3586 (2005).
- Beltowski J. Role of leptin in blood pressure regulation and arterial hypertension. J. Hypertens.24(5), 789–801 (2006).
- Straznicky NE, Lambert EA, Lambert GW, Masuo K, Esler MD, Nestel PJ. Effects of Dietary Weight Loss on Sympathetic Activity and Cardiac Risk Factors Associated with the Metabolic Syndrome. J. Clin. Endocrinol. Metab.90(11), 5998–6005 (2005).
- Oral EA, Simha V, Ruiz E et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med.346(8), 570–578 (2002).
- Banks WA, Coon AB, Robinson SM et al. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes53(5), 1253–1260 (2004).
- Lofgren IE, Herron KL, West KL et al. Weight loss favorably modifies anthropometrics and reverses the metabolic syndrome in premenopausal women. J. Am. Coll. Nutr.24(6), 486–493 (2005).
- Stiegler P, Cunliffe A. The role of diet and exercise for the maintenance of fat-free mass and resting metabolic rate during weight loss. Sports Med.36(3), 239–262 (2006).
- Chu NF, Stampfer MJ, Spiegelman D, Rifai N, Hotamisligil GS, Rimm EB. Dietary and lifestyle factors in relation to plasma leptin concentrations among normal weight and overweight men. Int. J. Obes. Relat. Metab. Disord.25(1), 106–114 (2001).
- Suviolahti E, Reue K, Cantor RM et al. Cross-species analyses implicate lipin 1 involvement in human glucose metabolism. Hum. Mol. Genet.15(3), 377–386 (2005).
- Kern PA. Potential role of TNFα and lipoprotein lipase as candidate genes for obesity. J. Nutr.127(9), 1917S–1922S (1997).
- Couillard C, Bergeron N, Prud’homme D et al. Postprandial triglyceride response in visceral obesity in men. Diabetes47(6), 953–960 (1998).
- Shoji T, Nishizawa Y, Koyama H et al. Lipoprotein metabolism in normolipidemic obese women during very low calorie diet: changes in high density lipoprotein. J. Nutr. Sci. Vitaminol. (Tokyo)37(Suppl.) S57–S64 (1991).
- Kern PA, Ong JM, Saffari B, Carty J. The effects of weight loss on the activity and expression of adipose-tissue lipoprotein lipase in very obese humans. N. Engl. J. Med.322(15), 1053–1059 (1990).
- Berman DM, Nicklas BJ, Ryan AS, Rogus EM, Dennis KE, Goldberg AP. Regulation of lipolysis and lipoprotein lipase after weight loss in obese, postmenopausal women. Obes. Res.12(1), 32–39 (2004).
- Patalay M, Lofgren IE, Freake HC, Koo SI, Fernandez ML. The lowering of plasma lipids following a weight reduction program is related to increased expression of the LDL receptor and lipoprotein lipase. J. Nutr.135(4), 735–739 (2005).
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation98(19), 2088–2093 (1998).
- Kolehmainen M, Vidal H, Ohisalo JJ, Pirinen E, Alhava E, Uusitupa MI. Hormone sensitive lipase expression and adipose tissue metabolism show gender difference in obese subjects after weight loss. Int. J. Obes. Relat. Metab. Disord.26(1), 6–16 (2002).
- Gesta S, Simon MF, Rey A et al. Secretion of a lysophospholipase D activity by adipocytes: involvement in lysophosphatidic acid synthesis. J. Lipid Res.43(6), 904–910 (2002).
- Moore HP, Silver RB, Mottillo EP, Bernlohr DA, Granneman JG. Perilipin targets a novel pool of lipid droplets for lipolytic attack by hormone-sensitive lipase. J. Biol. Chem.280(52), 43109–43120 (2005).
- Martinez-Botas J, Anderson JB, Tessier D et al. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat.Genet.26(4), 474–479 (2000).
- Wang Y, Sullivan S, Trujillo M et al. Perilipin expression in human adipose tissues: effects of severe obesity, gender, and depot. Obes. Res.11(8), 930–936 (2003).
- Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes44(8), 863–870 (1995).
- Corella D, Qi L, Sorli JV et al. Obese subjects carrying the 11482G>A polymorphism at the perilipin locus are resistant to weight loss after dietary energy restriction. J. Clin. Endocrinol. Metab.90(9), 5121–5126 (2005).
- Franckhauser S, Munoz S, Pujol A et al. Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes51(3), 624–630 (2002).
- Beale EG, Hammer RE, Antoine B, Forest C. Glyceroneogenesis comes of age. FASEB J.16(13), 1695–1696 (2002).
- Huuskonen J, Olkkonen VM, Jauhiainen M, Ehnholm C. The impact of phospholipid transfer protein (PLTP) on HDL metabolism. Atherosclerosis155(2), 269–281 (2001).
- Kaser S, Sandhofer A, Foger B et al. Influence of obesity and insulin sensitivity on phospholipid transfer protein activity. Diabetologia44(9), 1111–1117 (2001).
- Schlitt A, Bickel C, Thumma P et al. High plasma phospholipid transfer protein levels as a risk factor for coronary artery disease. Arterioscler. Thromb. Vasc. Biol.23(10), 1857–1862 (2003).
- Kaser S, Laimer M, Sandhofer A, Salzmann K, Ebenbichler CF, Patsch JR. Effects of weight loss on PLTP activity and HDL particle size. Int. J. Obes. Relat. Metab. Disord.28(10), 1280–1282 (2004).
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation106(25), 3143–3421 (2002).
- Lichtenstein AH, Ausman LM, Jalbert SM et al. Efficacy of a Therapeutic Lifestyle Change/Step 2 diet in moderately hypercholesterolemic middle-aged and elderly female and male subjects. J. Lipid Res.43(2), 264–273 (2002).
- Takamiya T, Kadowaki T, Zaky WR et al. The determinants of plasma plasminogen activator inhibitor-1 levels differ for American and Japanese men aged 40–49. Diabetes Res. Clin. Pract.72(2), 176–182 (2005).
- Festa A, D’Agostino R Jr, Tracy RP, Haffner SM. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes51(4), 1131–1137 (2002).
- Morange PE, Lijnen HR, Alessi MC, Kopp F, Collen D, Juhan-Vague I. Influence of PAI-1 on adipose tissue growth and metabolic parameters in a murine model of diet-induced obesity. Arterioscler. Thromb. Vasc. Biol.20(4), 1150–1154 (2000).
- Liang X, Kanjanabuch T, Mao SL et al. Plasminogen activator inhibitor-1 modulates adipocyte differentiation. Am. J. Physiol. Endocrinol. Metab.290(1), E103–E113 (2006).
- Hamalainen H, Ronnemaa T, Virtanen A et al. Improved fibrinolysis by an intensive lifestyle intervention in subjects with impaired glucose tolerance. The Finnish Diabetes Prevention Study. Diabetologia48(11), 2248–2253 (2005).
- Koerner A, Kratzsch J, Kiess W. Adipocytokines: leptin-the classical, resistin-the controversial, adiponectin-the promising, and more to come. Best Pract. Res. Clin. Endocrinol. Metab.19(4), 525–546 (2005).
- Yang Q, Graham TE, Mody N et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature436(7049), 356–362 (2005).
- Burri BJ, Neidlinger TR, Van Loan M, Keim NL. Effect of low-calorie diets on plasma retinol-binding protein concentrations in overweight women. J. Nutr. Biochem.1(9), 484–486 (1990).
- Gillmer MD. Mechanism of action/effects of androgens on lipid metabolism. Int. J. Fertil.37(Suppl. 2) 83–92 (1992).
- Niskanen L, Laaksonen DE, Punnonen K, Mustajoki P, Kaukua J, Rissanen A. Changes in sex hormone-binding globulin and testosterone during weight loss and weight maintenance in abdominally obese men with the metabolic syndrome. Diabetes Obes. Metab.6(3), 208–215 (2004).
- Kaukua J, Pekkarinen T, Sane T, Mustajoki P. Sex hormones and sexual function in obese men losing weight. Obes. Res.11(6), 689–694 (2003).
- Stanik S, Dornfeld LP, Maxwell MH, Viosca SP, Korenman SG. The effect of weight loss on reproductive hormones in obese men. J. Clin. Endocrinol. Metab.53(4), 828–832 (1981).
- Strain GW, Zumoff B, Miller LK et al. Effect of massive weight loss on hypothalamic-pituitary-gonadal function in obese men. J. Clin. Endocrinol. Metab.66(5), 1019–1023 (1988).
- Pasquali R, Casimirri F, Melchionda N et al. Weight loss and sex steroid metabolism in massively obese man. J. Endocrinol. Invest.11(3), 205–210 (1988).
- Escobar-Morreale HF, Botella-Carretero JI, varez-Blasco F, Sancho J, San Millan JL. The polycystic ovary syndrome associated with morbid obesity may resolve after weight loss induced by bariatric surgery. J. Clin. Endocrinol. Metab.90(12), 6364–6369 (2005).
- Hoeger K. Obesity and weight loss in polycystic ovary syndrome. Obstet. Gynecol. Clin. North Am.28(1), 85–97 (2001).
- Zhang S, Kim KH. TNF-α inhibits glucose-induced insulin secretion in a pancreatic β-cell line (INS-1). FEBS Lett.377(2), 237–239 (1995).
- Berndt J, Kloting N, Kralisch S et al. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes54(10), 2911–2916 (2005).
- Kloting N, Berndt J, Kralisch S et al. Vaspin gene expression in human adipose tissue: Association with obesity and type 2 diabetes. Biochem. Biophys. Res. Commun.339(1), 430–436 (2006).
- Hida K, Wada J, Eguchi J et al. Visceral adipose tissue-derived serine protease inhibitor: a unique insulin-sensitizing adipocytokine in obesity. Proc. Natl Acad. Sci. USA102(30), 10610–10615 (2005).
- Chen MP, Chung FM, Chang DM et al. Elevated Plasma Level of Visfatin/Pre-B Cell Colony-Enhancing Factor in Patients with Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab.91(1), 295–299 (2006).
- Haider DG, Schindler K, Schaller G, Prager G, Wolzt M, Ludvik B. Increased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric banding. J. Clin. Endocrinol. Metab.91(4), 1578–1581 (2006).
- Bays HE, Dujovne CA. Adiposopathy is a more rational treatment target for metabolic disease than obesity alone. Curr. Atheroscler. Rep. (In Press) (2006).
- Bays H, Stein EA. Pharmacotherapy for dyslipidaemia–current therapies and future agents. Expert Opin. Pharmacother.4(11), 1901–1938 (2003).
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet365(9468), 1389–1397 (2005).
- Hsieh CJ, Wang PW, Liu RT et al. Orlistat for obesity: benefits beyond weight loss. Diabetes Res. Clin. Pract.67(1), 78–83 (2005).
- Bays HE. The melanocortin system as a therapeutic treatment target for adiposity and adiposopathy. Drugs in R & D (In Press) (2006).
- Bartness TJ, Kay Song C, Shi H, Bowers RR, Foster MT. Brain-adipose tissue cross talk. Proc. Nutr. Soc.64(1), 53–64 (2005).
- Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol.277(6 Pt 1), E1130–E1141 (1999).
- Blaak EE, Wolffenbuttel BH, Saris WH, Pelsers MM, Wagenmakers AJ. Weight reduction and the impaired plasma-derived free fatty acid oxidation in type 2 diabetic subjects. J. Clin. Endocrinol. Metab.86(4), 1638–1644 (2001).
- Kelley DE Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a re-examination. Diabetes49677–683 (2000).
- Tomas F, Kelly M, Xiang X et al. Metabolic and hormonal interactions between muscle and adipose tissue. Proc. Nutr. Soc.63(2), 381–385 (4).
- Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition22(7–8), 830–844 (2006).
- Chu CA, Sherck SM, Igawa K et al. Effects of free fatty acids on hepatic glycogenolysis and gluconeogenesis in conscious dogs. Am. J. Physiol. Endocrinol. Metab.282(2), E402–E411 (2002).
- Arner P. Regional adipocity in man. J. Endocrinol.155(2), 191–192 (1997).
- Wajchenberg BL, Giannella-Neto D, da Silva ME, Santos RF. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm. Metab. Res.34(11–12), 616–621 (2002).
- Park KG, Park KS, Kim MJ et al. Relationship between serum adiponectin and leptin concentrations and body fat distribution. Diabetes Res. Clin. Pract.63(2), 135–142 (2004).
- Pedersen SB, Kristensen K, Hermann PA, Katzenellenbogen JA, Richelsen B. Estrogen controls lipolysis by up-regulating α2A-adrenergic receptors directly in human adipose tissue through the estrogen receptor α. Implications for the female fat distribution. J. Clin. Endocrinol. Metab.89(4), 1869–1878 (2004).
- McCarty MF. A paradox resolved: the postprandial model of insulin resistance explains why gynoid adiposity appears to be protective. Med. Hypotheses61(2), 173–176 (2003).
- Reynisdottir S, Dauzats M, Thorne A, Langin D. Comparison of hormone-sensitive lipase activity in visceral and subcutaneous human adipose tissue. J. Clin.Endocrinol.Metab.82(12), 4162–4166 (1997).
- Mohamed-Ali V, Goodrick S, Rawesh A et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-α, in vivo. J. Clin.Endocrinol.Metab.82(12), 4196–4200 (1997).
- Raz I, Eldor R, Cernea S, Shafrir E. Diabetes: insulin resistance and derangements in lipid metabolism. Cure through intervention in fat transport and storage. Diabetes Metab. Res. Rev.21(1), 3–14 (2005).
- Rieusset J, Auwerx J, Vidal H. Regulation of gene expression by activation of the peroxisome proliferator-activated receptor γ with rosiglitazone (BRL 49653) in human adipocytes. Biochem. Biophys. Res. Commun.265(1), 265–271 (1999).
- Ebeling P, Teppo AM, Koistinen HA, Koivisto VA. Concentration of the complement activation product, acylation-stimulating protein, is related to C-reactive protein in patients with type 2 diabetes. Metabolism50(3), 283–287 (2001).
- Bailey CJ. Treating insulin resistance in type 2 diabetes with metformin and thiazolidinediones. Diabetes Obes. Metab.7(6), 675–691 (2005).
- Diez JJ, Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur. J. Endocrinol.148(3), 293–300 (2003).
- Filippatos TD, Kiortsis DN, Liberopoulos EN, Mikhailidis DP, Elisaf MS. A review of the metabolic effects of sibutramine. Curr. Med. Res. Opin.21(3), 457–468 (2005).
- Hung YJ, Chen YC, Pei D et al. Sibutramine improves insulin sensitivity without alteration of serum adiponectin in obese subjects with Type 2 diabetes. Diabet. Med.22(8), 1024–1030 (2005).
- Despres JP, Golay A, Sjostrom L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med.353(20), 2121–2134 (2005).
- Wu Z, Xie Y, Morrison RF, Bucher NL, Farmer SR. PPAR-γ induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPα during the conversion of 3T3 fibroblasts into adipocytes. J. Clin. Invest.101(1), 22–32 (1998).
- Unger RH. Lipotoxic diseases. Annu. Rev. Med.53, 319–336 (2002).
- van Tits LJ, Rioglu-Oral E, Sweep CG, Smits P, Stalenhoef AF, Tack CJ. Anti-inflammatory effects of troglitazone in nondiabetic obese subjects independent of changes in insulin sensitivity. Neth. J. Med.63(7), 250–255 (2005).
- Yesilbursa D, Serdar A, Heper Y et al. The effect of orlistat-induced weight loss on interleukin-6 and C-reactive protein levels in obese subjects. Acta Cardiol.60(3), 265–269 (2005).
- Kletzien RF, Clarke SD, Ulrich RG. Enhancement of adipocyte differentiation by an insulin-sensitizing agent. Mol. Pharmacol.41(2), 393–398 (1992).
- Drent ML, Popp-Snijders C, Ader HJ, Jansen JB, van der V. Lipase inhibition and hormonal status, body composition and gastrointestinal processing of a liquid high-fat mixed meal in moderately obese subjects. Obes. Res.3(6), 573–581 (1995).
- Baranowska B, Wolinska-Witort E, Martynska L et al. Sibutramine therapy in obese women - effects on plasma neuropeptide Y (NPY), insulin, leptin and β-endorphin concentrations. Neuro. Endocrinol. Lett.26(6), 675–679 (2005).
- Poirier B, Bidouard JP, Cadrouvele C et al. The anti-obesity effect of rimonabant is associated with an improved serum lipid profile. Diabetes Obes. Metab.7(1), 65–72 (2005).
- Bouaboula M, Hilairet S, Marchand J, Fajas L, Le Fur G, Casellas P. Anandamide induced PPARγ transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol.517(3), 174–181 (2005).
- Simon MF, Daviaud D, Pradere JP et al. Lysophosphatidic acid inhibits adipocyte differentiation via lysophosphatidic acid 1 receptor-dependent down-regulation of peroxisome proliferator-activated receptor γ2. J. Biol. Chem.280(15), 14656–14662 (2005).
- Rissanen P, Vahtera E, Krusius T, Uusitupa M, Rissanen A. Weight change and blood coagulability and fibrinolysis in healthy obese women. Int. J. Obes. Relat. Metab. Disord.25(2), 212–218 (2001).
- Jung HS, Youn BS, Cho YM et al. The effects of rosiglitazone and metformin on the plasma concentrations of resistin in patients with type 2 diabetes mellitus. Metabolism54(3), 314–320 (2005).
- Costa B, Trovato AE, Colleoni M, Giagnoni G, Zarini E, Croci T. Effect of the cannabinoid CB1 receptor antagonist, SR141716, on nociceptive response and nerve demyelination in rodents with chronic constriction injury of the sciatic nerve. Pain116(1–2), 52–61 (2005).
- Hammarstedt A, Pihlajamaki J, Sopasakis VR et al. Visfatin is an adipokine but it is not regulated by thiazolidinediones. J. Clin. Endocrinol. Metab.91(3), 1181–1184 (2006).
- Choi KC, Ryu OH, Lee KW et al. Effect of PPAR-α and -γ agonist on the expression of visfatin, adiponectin, and TNF-α in visceral fat of OLETF rats. Biochem. Biophys. Res. Commun.336(3), 747–753 (2005).
- Visser L, Zuurbier CJ, van Wezel HB, van der Vusse GJ, Hoek FJ. Overestimation of plasma nonesterified fatty acid concentrations in heparinized blood. Circulation110(13), E328 (2004).
- Diep QN, El Mabrouk M, Cohn JS et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-γ. Circulation105(19), 2296–2302 (2002).
- Cota D, Marsicano G, Tschop M et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Invest.112(3), 423–431 (2003).
- Dereli D, Dereli T, Bayraktar F, Ozgen AG, Yilmaz C. Endocrine and metabolic effects of rosiglitazone in non-obese women with polycystic ovary disease. Endocr. J.52(3), 299–308 (2005).
- Vierhapper H, Nowotny P, Waldhausl W. Reduced production rates of testosterone and dihydrotestosterone in healthy men treated with rosiglitazone. Metabolism52(2), 230–232 (2003).
- Jayagopal V, Kilpatrick ES, Holding S, Jennings PE, Atkin SL. Orlistat is as beneficial as metformin in the treatment of polycystic ovarian syndrome. J. Clin. Endocrinol. Metab.90(2), 729–733 (2005).
- Sabuncu T, Harma M, Harma M, Nazligul Y, Kilic F. Sibutramine has a positive effect on clinical and metabolic parameters in obese patients with polycystic ovary syndrome. Fertil. Steril.80(5), 1199–1204 (2003).
- Pang XH, Gu W. [Association of serum sex hormone-binding globulin with type 2 diabetes]. Zhejiang. Da. Xue. Xue. Bao. Yi. Xue. Ban.33(1), 60–64 (2004).
- Rubin GL, Zhao Y, Kalus AM, Simpson ER. Peroxisome proliferator-activated receptor γ ligands inhibit estrogen biosynthesis in human breast adipose tissue: possible implications for breast cancer therapy. Cancer Res.60(6), 1604–1608 (2000).
Websites
- Bays H. Adiposopathy causes the metabolic syndrome: the beginning or end of a controversy? IAS commentary. Home of the International Atherosclerosis Society www.athero.org
- Bays H. Adiposopathy homepage. www.lmarc.com/Index%20Adiposopathy%20Frames.htm