Abstract
Hydrogen sulfide is rapidly gaining ground as a physiological mediator of inflammation, but there is no clear consensus as to its precise role in inflammatory signaling. This article discusses the disparate anti-inflammatory (‘the good’) and proinflammatory (‘the bad’) effects of endogenous and pharmacological H2S in disparate animal model and cell culture systems. We also discuss ‘the ugly’, such as problems of using wholly specific inhibitors of enzymatic H2S synthesis, and the use of pharmacological donor compounds, which release H2S too quickly to be physiologically representative of endogenous H2S synthesis. Furthermore, recently developed slow-release H2S donors, which offer a more physiological approach to understanding the complex role of H2S in acute and chronic inflammation (‘the promising’) are discussed.
Plasma and synovial fluid aspirates were obtained from healthy controls, patients with OA (n = 5), RA (n = 27), ReA (n = 7) and PsA (n = 5) and H2S levels determined by zinc-trap spectrophotometry, as described in Citation[18,22]. All patients and volunteers in this study were enrolled following institutional ethical approval (North Devon and Exeter Research Ethics Committee #04/Q2102/87 and #05/Q2103/91). All patients with RA and OA fulfilled the American College of Rheumatology criteria Citation[143] and attended the department of Rheumatology, Royal Devon and Exeter Hospital Trust and had given informed written consent. Data are shown as mean ± standard deviation. Statistical analysis was by ANOVA.
H2S: Hydrogen sulfide; OA: Osteoarthritis; PsA: Psoriatic arthritis; RA: Rheumatoid arthritis; ReA: Reactive arthritis.
![Figure 1. Comparison of synovial fluid concentrations of H2S in inflammatory and noninflammatory arthritides.Plasma and synovial fluid aspirates were obtained from healthy controls, patients with OA (n = 5), RA (n = 27), ReA (n = 7) and PsA (n = 5) and H2S levels determined by zinc-trap spectrophotometry, as described in Citation[18,22]. All patients and volunteers in this study were enrolled following institutional ethical approval (North Devon and Exeter Research Ethics Committee #04/Q2102/87 and #05/Q2103/91). All patients with RA and OA fulfilled the American College of Rheumatology criteria Citation[143] and attended the department of Rheumatology, Royal Devon and Exeter Hospital Trust and had given informed written consent. Data are shown as mean ± standard deviation. Statistical analysis was by ANOVA.H2S: Hydrogen sulfide; OA: Osteoarthritis; PsA: Psoriatic arthritis; RA: Rheumatoid arthritis; ReA: Reactive arthritis.](/cms/asset/15efd026-8198-4db7-8839-4bc52ce38e77/ierj_a_11209390_f0001_b.jpg)
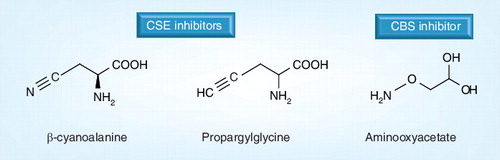
Inhibitors of CSE: dL-propargylglycine and β-cyanoalanine. Inhibitor of CBS: aminooxyacetate.
CBS: Cystathionine β-synthase; CSE: Cystathionine γ-lyase.
ADT-OH: 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione; H2S: Hydrogen sulfide.
Inflammation
Inflammation is a protective physiological reaction of vascularized tissue to local injury or tissue destruction Citation[1–3]. In its broadest sense, inflammation is the host response to tissue injury to remove injurious stimuli and initiate a healing process. It is a complex network of coordinated cellular responses designed to destroy, dilute or ‘wall-off’ both the noxious stimuli and the injured tissue Citation[3,4]. The five clinical (‘cardinal’) signs of inflammation are redness (rubor), heat (calor), swelling (tumor), pain (dolor) and eventually loss of function (functio laesa) Citation[2,3]. These cardinal signs represent the macroscopic culmination of a multitude of molecular and cellular processes, many of which have become well defined over the last century, and may be reproduced in convenient in vitro experimental systems or in more complex and, perhaps more importantly, in relevant animal models in vivo. Although detailed descriptions of the myriad inflammatory pathways are the subject of a plethora of review articles and medical text books, for the context of this current article, a brief discussion of the general inflammatory process is pertinent.
The inflammatory response is often categorized according to the duration and kinetics of the reaction and can be classified as either acute or chronic Citation[2,3]. In general terms, acute inflammation is the immediate and early host response to an injurous agent. It is mediated by an increase in blood flow into injured tissues primarily from arteriolar dilatation and the opening of capillary beds. There is also increased movement of leukocytes (in particular granulocytes, such as neutrophils) and increased leukocyte adhesion to the vascular endothelium (via adhesion molecules). Leukocytes phagocytose the injurious agent and release toxic metabolites and proteases, which potentially causes tissue damage. These processes result in a cascade of biochemical mediators, such as vasoactive amines (e.g., histamine and 5-hydroxytryptamine), plasma proteases (e.g., the complement, kinin and clotting systems) and arachidonic acid metabolites (e.g., prostaglandins, leukotrienes and lipoxins) Citation[5]. A complex array of cytokines and chemokines are also produced by inflammatory cells and the vascular endothelium, which regulate lymphocyte function through activating (e.g., IL-2, IL-4, IFN-α and IFN-β) or inhibiting (e.g., IL-10 and TGF-β) immune responses. Cytokines activate inflammatory cells (e.g., TNF-α, IL-1β, IFN-γ and IL-6) or stimulate hematopoiesis and leukocyte growth and differentiation (e.g., IL-3 and IL-7) Citation[5]. Physiological gaseous mediators, such as nitric oxide (•NO) and carbon monoxide (CO; reviewed in explicit detail elsewhere Citation[6–10]) have been proposed to induce, inhibit and regulate the inflammatory process.
More recently, a third endogenous gas, hydrogen sulfide (H2S), has been proposed as an additional inflammatory mediator Citation[11], although the precise role for H2S is controversial (see later; detailed discussion on endogenous synthesis of H2S is contained elsewhere in this article). Therefore, acute inflammation manifests as vascular changes, edema and largely neutrophillic infiltration. Examples of acute inflammation are septic shock, multiple organ failure and pancreatitis.
Chronic inflammation, on the other hand, is difficult to define precisely. In general terms, it is inflammation of prolonged duration (weeks or months), in which active inflammation, tissue destruction and attempts at repair proceed simultaneously Citation[2], and leads to several distinct processes:
• A progressive shift in the types of cells present at the site of inflammation (e.g., from granulocytes to mononuclear cells, such as macrophages, lymphocytes and plasma cells, which characterize the persistent reaction to injury);
• Tissue destruction mediated predominantly by the inflammatory cells;
• Connective tissue replacement of damaged tissue via microvascular proliferation (angiogenesis) and fibrosis (attempts at healing).
Although chronic inflammation may follow acute inflammation, frequently, it has insidious beginnings as a simmering low-grade and often asymptomatic response, so-called ‘subclinical inflammation’. Types of chronic inflammation include some of the most common and debilitating human diseases, such as rheumatoid arthritis, tuberculosis, asthma, inflammatory bowel disease, vasculitis and Crohn’s disease. Consequently, pharmacological agents to treat these disorders represent a multibillion dollar industry and novel chemical entities, which prevent, inhibit or resolve inflammation, are of continual interest to the global pharmaceutical industry.
Recent evidence for a role of H2S in inflammation
H2S synthesis & biochemistry
The synthesis pathways and biochemistry of H2S are reviewed extensively throughout this article. However, a generalized overview to familarize the reader with the topic is pertinent. H2S has been proposed as a third physiological gaseous mediator along with CO and •NO, for which there appears to be considerable interplay (reviewed elsewhere Citation[6,12]). Its formation in vivo is catalyzed predominantly by the pyridoxal phosphate-dependent enzymes cystathionine-γ-lyase (CSE) and cystathionine β-synthase (CBS), utilizing the amino acids l-cysteine, l-homocysteine and l-cystathionine (reviewed in Citation[12]). In the last decade, H2S has been shown to regulate a steadily growing list of physiological functions, such as blood pressure Citation[13], insulin secretion Citation[14,15], nociception Citation[16], learning and memory Citation[17] and inflammation Citation[6], in part via the modulation of KATP channel activity. As such, modulation of endogenous H2S through either inhibitors of its enzymatic synthesis or H2S-donor molecules represent viable and potentially therapeutic targets for a variety of human diseases where perturbed H2S synthesis is involved, such as hypertension Citation[13,18,19], diabetes and obesity Citation[18], hemorrhagic Citation[20], endotoxic shock Citation[21] and chronic inflammation (e.g., arthritis Citation[22] and inflammatory bowel disease Citation[23].
However, as we will discuss, the precise role for H2S is controversial, and as with any new and emerging field of research, progress has been hampered by the limitations on selective tools, such as CSE and CBS inhibitors and H2S-donor molecules that model endogenous enzymatic H2S synthesis. It should be noted that H2S is a weak acid (pKa = 6.96), and at physiological pH 7.4, H2S will always exist as approximately a third H2S and two-thirds H+ and HS- (hydrosulfide anion). At present, it is unknown whether the biological effects of H2S are mediated directly by H2S or by HS-, or whether a combination of both species is required. During the inflammatory process, mildly acidic conditions may predominate, which would shift the equilibrium towards H2S. Therefore, we prudently use the term ‘H2S’ to encompass H2S and HS-.
H2S & acute whole-body inflammation (sepsis)
Sepsis and its sequelae (e.g., septic shock and multiple organ failure) are the most common cause of death in medical and surgical intensive units Citation[24]. The predominant view of sepsis is one of acute whole-body inflammation in the presence of a known or suspected infection, most commonly caused by bacteria, but also induced by fungi, viruses and protozoa Citation[2,24], resulting in massively elevated levels of inflammatory markers (e.g., acute-phase proteins, cytokines and NO and prostanoids). This results in tissue hypoperfusion and organ dysfunction (severe sepsis) and, in septic shock, persistent hypotension Citation[2]. Both hypoperfusion and hypotension damage the vascular endothelium and impair tissue respiration leading to high mortality rates – 16% in sepsis to up to 60% in patients with septic shock Citation[25].
Bacterial endotoxins, such as lipopolysaccharide (LPS), have previously been shown to induce excessive activation and upregulation of vascular KATP channels, to induce hypotension and to substantially reduce vascular sensitivity to vasoconstrictive agents Citation[26,27]. H2S has been proposed as a potential endogenous ligand for KATP channels and induces KATP channel-mediated vasorelaxation Citation[28] in several vascular tissues, promoting speculation that H2S may play a role in endotoxic shock, an important form of acute inflammation, as mentioned. Indeed, early studies using cecal ligation and puncture (CLP) or injection of bacterial LPS into rats or mice significantly increased the expression of CSE and CBS and H2S synthesis in plasma, vascular tissues, lung, liver, kidney and pancreas Citation[21,29–32]. This suggested that, as with •NO synthesis (via inducible nitric oxide synthase [iNOS]), H2S synthesis is also an inducible phenomenon. Plasma H2S levels in patients with septic shock were also reported to be elevated up to four-times compared with control subjects (up to 200 µmol/l in one patient) Citation[21]. At these plasma concentrations, H2S induces KATP-dependent vasodilatation in anesthetized animals and in isolated aortic rings Citation[28,33,34]. Conversely, administration of the CSE inhibitor propargylglycine (PAG; see later) to LPS- or CLP-treated animals decreased plasma H2S levels significantly, tissue edema and increased survival Citation[21,29,30], suggesting that elevated plasma H2S was likely to be associated with the hemodynamic collapse observed in septic shock Citation[21]. It should be noted that the protective effects of PAG may be independent of the improvement of hemodynamics (or CSE), since PAG treatment alone did not alter systemic blood pressure or affect LPS-induced hypotension but did significantly reduce the hepatic, pancreatic and neuromuscular damage induced by LPS Citation[31]. In hemorrhagic shock however, blood withdrawal led to significantly increased liver (but not kidney) and plasma levels of H2S, and pretreatment of rats with PAG or β-cyanoalanine (BCA; a reversible inhibitor of CSE) reduced liver H2S synthesis and partially restored systemic blood pressure Citation[20,35].
A role for H2S in the acute inflammatory aspects of septic and hemorrhagic shock is also evident. For example, PAG treatment of LPS- and CLP-treated animals decreased lung and liver myeloperoxidase (MPO) expression and activity, ameliorated histological lung and hepatic damage and lowered plasma levels of TNF-α and nitrite/nitrate (an index of •NO synthesis) Citation[21,29–31]. Similarly, pretreatment of rats with PAG for 1 h prior to blood withdrawal was also protective as PAG increased heart rate recovery time, significantly reduced liver, lung and plasma levels of TNF-α and IL-6 and, in the liver, PAG reduced neutrophilic accumulation and tissue damage Citation[35]. Conversely, administration of H2S donors based on sulfide salts (see later), such as sodium hydrosulfide (NaSH) to animals either alone or in combination with LPS, induce marked hypotension, liver, lung and kidney inflammation and aggravated multiple organ injury through upregulation of tissue NF-κB, p38 and ERK1/2 signaling Citation[36–38].
In summary, the aforementioned studies suggest that in septic and hemorrhagic shock, induction of endogenous H2S synthesis is detrimental, since PAG and BCA (CSE inhibitors; see later) reduced inflammation and multiple organ dysfunction in septic shock, and were protective in hemorrhagic shock, and the H2S-donor NaSH promoted inflammation and tissue injury.
H2S & inflammatory swelling (edema)
Edema is the excessive accumulation of serous fluid in the intercellular spaces of tissues in skin or in one or more cavities of the body, which occurs as part of the acute inflammatory response because of increased blood vessel wall permeability. Inhalation of H2S gas is known to induce substantive pulmonary edema, suggesting endogenous H2S could also promote tissue swelling. Edema is commonly modeled in the laboratory through the use of carrageenan, a mucopolysaccharide extract of Chondrus crispus that induces an acute, defined and nonimmune inflammatory response Citation[39]. Injection of carrageenan into the hindpaw of rats induced a significant increase in H2S synthesis and neutrophil infiltration, whereas pretreatment with PAG (a CSE inhibitor) resulted in dose-dependent inhibition of hindpaw edema and MPO activity Citation[40]. In sharp contrast to this, a more recent study Citation[41] showed the H2S ‘donors’ NaSH, sodium sulfide (Na2S) and Lawesson’s reagent reduced leukocyte infiltration and adherence to the vascular endothelium via glibenclamide-inhibitable KATP channels, and the CSE inhibitor BCA promoted leukocyte infiltration. However, the effects of PAG on hindpaw H2S levels were not determined in this study Citation[41]. A closer examination of the experimental protocol used in both studies suggests inter-laboratory methodological differences could have accounted for these apparent contradictions. For example, Bhatia et al. treated rats with 2% w/v solution of carrageenan for 3 h and added PAG intraplantar into one hindpaw 1 h before carrageenan treatment Citation[40], whereas Zanardo et al. performed time-course studies and used a 1.5% w/v solution, injecting BCA (or H2S donors) intraperitoneally 30 min prior to carrageenan treatment Citation[41]. It is also possible that BCA and/or PAG exhibited effects independent of CSE inhibition, which modulated hindpaw fluid homeostasis (see later). Therefore, at present, the precise role of H2S in mediating edema is unresolved.
Role of H2S in gastrointestinal inflammation
The class of drugs known as NSAIDs, such as aspirin, diclofenac, ibuprofen and celecoxib, are among the most widely prescribed medications in the world and, owing to their antipyretic, analgesic and anti-inflammatory properties, they are used in the treatment of a wide range of conditions, such as headache, migraine, rheumatoid arthritis, osteoarthritis, acute gout, pyrexia and dysmenorrhea. Their primary mode of action is the inhibition of prostaglandin production from the enzymes COX-1 and -2. In the gastric mucosa, prostaglandins (PGs), such as PGE2, PGI2, PGF2a and PGD2, mediate many of the components of ‘gastric mucosal defense’ Citation[42,43] and maintain gastric blood flow, regulate the secretion of bicarbonate and mucus by surface epithelial cells and repair superficial mucosal injury through epithelial restitution Citation[42,43]. However, while inhibition of COX activity is useful systemically in reducing pain, edema and inflammation, inhibition of COX-1-mediated prostaglandin synthesis in the gastric mucosa impairs blood flow and induces superficial injury. This subsequently triggers an acute inflammatory response, ultimately leading to gastric ulceration (explicitly reviewed in Citation[43]) in 1–4% of patients taking NSAIDs chronically Citation[44].
Recently, modulation of endogenous gastric H2S synthesis has been suggested as one potential mechanism by which NSAIDs induce gastrotoxicity Citation[45]. CSE and CBS are variably expressed throughout the stomach, jejunum and colon of rats and mice, and low levels of CSE and CBS are present in human colon biopsies, and experiments with CSE and CBS inhibitors showed both enzymes to readily synthesized H2S Citation[23]. Further immunohistochemical studies showed CBS immunostaining was primarily localized to cells within the muscularis mucosa, submucosa, lamina propria and fibroblasts, whereas CSE staining was more diffuse, possibly indicating an association with blood vessels Citation[23,46]. NSAIDs reduced the expression of CSE and H2S production in the gastric mucosa, induced histological lesions Citation[41,45] and potentiated acetic acid-induced gastric ulcer formation Citation[47]. Conversely, NaSH induced gastroprotective effects, such as reducing NSAID-induced adherence of leukocytes to the vascular endothelium and reducing mucosal neutrophil infiltration. NaSH also decreased the expression of TNF-α, intercellular adhesion molecule 1 (ICAM-1) and lymphocyte-associated antigen-1, but no significant effect on mucosal PGE2 synthesis was observed Citation[45]. Interestingly, the KATP channel antagonist glibenclamide and the CSE inhibitor PAG (see later) aggravated NSAID-induced gastric injury, whereas pinacidil (a KATP channel agonist) reduced gastric injury Citation[45]. Although l-cysteine enhanced ulcer healing, an effect prevented by PAG, this was not inhibited by glibenclamide, suggesting that the presence of protective molecular pathways is independent of KATP channel activation in this model Citation[47].
Impaired synthesis of H2S may also play a role in the development of inflammatory bowel disease and, in particular, ulcerative colitis, an idiopathic chronic inflammatory disorder limited to the colon that manifests as painful colonic ulcers. Recently, in the trinitrobenzenesulfonic acid (TNBS) rat model, Wallace et al., showed that during the development of colitis, levels of colonic H2S synthesis initially increased in response to TNBS, then decreased in parallel to colonic damage where H2S synthesis in the colon was mediated by both CSE and CBS enzymes Citation[46]. Furthermore, pharmacological inhibition of CBS and, to a lesser extent, CSE inhibition exacerbated colitis. By contrast, intracolonic administration of NaSH or Lawesson’s reagent (an additional H2S donor) decreased TNBS-induced colonic tissue damage and thickness and lowered TNF-α expression. Furthermore, treatment of rats with aminooxyacetic acid (AOAA), PAG and the KATP channel antagonist glibenclamide substantially increased mortality rates but this was not observed with the BCA (an additional CSE inhibitor) or pinacidil. The disparate effects of PAG and BCA could possibly be due to dosing, the fact that PAG binds to CSE irreversibly Citation[48], whereas BCA binding is reversible Citation[49], or that PAG (and AOAA) may induce effects independent of CSE inhibition (see later). However, collectively, these studies strongly suggest a potential role of H2S in regulating gastric inflammatory response and microvascular blood flow via KATP channel activation.
H2S & pancreatic inflammation
Both CSE and CBS are highly expressed in pancreatic acinar cells under basal conditions Citation[50]. Upon experimental induction of acute pancreatitis with the decapeptide secretagogue cerulein, acinar cell and lung H2S synthesis and CSE expression were significantly increased whereas CBS levels were reduced, suggesting CSE as the predominant source of H2S in this model Citation[50]. Cerulein also elevated plasma H2S levels. Inhibition of CSE in acinar cells Citation[50] or in cerulean-treated mice Citation[51] with PAG treatment reduced cerulein-induced formation of H2S in the lung and pancreas and increased expression of the macrophage chemokines MCP-1, MIP-1a and MIP-2, as well as significantly suppressing the proinflammatory neuropeptides substance P (SP), preprotachykinin-A (PPT-A) and neurokinin-1 receptor (NK1R), mediated, at least in part, by the activation of the transcription factor NF-κB Citation[52]. By contrast, the exposure of isolated acinar cells to NaSH (10–100 µmol/l) induced the synthesis of SP, PPT-A and NK1A Citation[50], activated the SFK and caused an upregulation of ICAM-1 Citation[52].
Collectively, the aforementioned studies have strongly suggested that H2S is proinflammatory in acute pancreatitis. However, the effects of H2S on the exocrine pancreas are complex. For example, in addition to stimulating the synthesis of proinflammatory mediators, NaSH also induces acinar cell apoptosis (1–100 µmol/l) through JNK/p38 and mitochondrial-dependent pathways Citation[53,54], suggesting that the aforementioned effects on proinflammatory signaling could be due to cell death. Low doses of NaSH were recently shown to reduce cerulein-induced neutrophil accumulation and histological tissue damage in the pancreas and lung, with concomitant downregulation of the pancreatic and pulmonary cell adhesion molecules P-selectin, E-selectin, ICAM-1 and VCAM-1 Citation[55]. The precise reasons for this discrepancy are unknown.
H2S & skin inflammation (burn injury)
To date, two studies reporting conflicting results have described a potential role for H2S in acute burn injury using anesthetized mice subjected to either 30 Citation[56,57] or 40% Citation[57] total body surface area (TBSA) full thickness burn injury. TBSA produces a large systemic inflammatory reaction characterized by leukocyte activation and plasma leakage in the microvasculature of tissues and organs remote from the wound. The degree of inflammatory response correlates directly with the percentage of TBSA burn Citation[58,59]. In mice subjected to 30% TBSA burn injury Citation[56,57], significantly elevated plasma and hepatic H2S levels with a concomitant increase in liver and lung expression of CSE were observed 8 h post-burn injury. In this study, prophylactic and therapeutic administration of PAG reduced burn-associated neutrophil accumulation and histological changes in liver and lung tissues. Injection of NaSH (10 mg/kg intraperitoneally) at the same time as burn injury aggravated burn-associated inflammation to a small, but significant, extent, suggesting H2S promoted tissue damage and inflammation. By sharp contrast, in animals receiving a 40% TBSA burn injury and smoke inhalation, administration of Na2S (2 mg/kg subcutaneously) immediately after injury increased survival, decreased lung oxidative stress and substantially reduced lung levels of the proinflammatory cytokine IL-1β but increased lung levels of the anti-inflammatory mediator IL-10 Citation[57]. However, the effects of PAG were not investigated in this study. The precise reasons for these disparate findings are unclear. In one study, Na2S was administered as a pH- and osmolarity-balanced solution Citation[57], whereas in the other study, NaSH was used but the disparity was unlikely to be due to the donor employed, since once administered to the animals they would give approximately equal total H2S Citation[55]. However, the route of administration, either intraperitoneal Citation[56] or subcutaneous Citation[57], could reflect significant differences in H2S bioavailability at the local sites of inflammation, which, in turn, might modulate local inflammatory signaling. A further possibility is an additive effect with subcutaneously injected analgesics. For example, buprenorphine was injected subcutaneously in the study by Esechie et al.Citation[57], while no analgesic was adminstered in the study by Zhang et al.Citation[56]. In summary, the role for H2S in burn injury is unclear.
H2S & inflammatory lung disease
Acute respiratory distress syndrome (ARDS) and the less-severe acute lung injury (ALI) are serious reactions to various forms of injuries to the lung (e.g., alcohol or drug abuse, trauma and burns) or acute illness (e.g., sepsis, acute pancreatitis and pneumonia). ARDS is characterized by inflammation of the lung parenchyma leading to impaired gas exchange, release of inflammatory mediators, inflammation, hypoxia, shortness of breath, tachypnea and often multiple organ failure Citation[2]. In the oleic acid (OA) model of ALI, OA decreased arterial PaO2, increased ALI disease score (index of quantitative assessment [IQA]), polymorphonuclear cell infiltration in the lungs and increased plasma and lung levels of H2S, IL-6, IL-8 and IL-10. By sharp contrast, injection of NaSH (56 µmol/l) 30 min prior to OA significantly reduced the extent of lung injury, lowered lung and plasma IL-8 and IL-6 levels and reduced inflammatory cell accumulation within 2 h. NaSH also increased lung and plasma IL-10 levels. Although no clear mechanisms were addressed and effects of CSE and CBS inhibitors were not investigated, it is clear that lower plasma (or lung) H2S levels were indicative of lung injury in this model, and strategies to raise lung H2S levels may be beneficial in ALI and ARDS.
In addition to a potential role in acute inflammatory conditions as summarized above, a role for H2S in chronic inflammation is emerging. H2S gas inhalation is well known to induce substantive pulmonary edema characterized by extensive eosinophilic extravasation into the bronchoalveolar space and fibrinocellular alveolitis Citation[59,60], suggesting modulation of endogenous H2S synthesis could represent a novel pathway in chronic inflammatory lung disease. Chronic obstructive pulmonary disease (COPD) is characterized by chronic inflammation throughout the airways, parenchymal and pulmonary vasculature, resulting in substantial airflow obstruction. Using a sulfide-specific electrode, Chen et al. showed significantly higher serum H2S levels in patients with COPD compared with age-matched control subjects or patients with acute exacerbation of COPD Citation[61,62]. Levels of H2S decreased with progressively worsening respiratory failure. In these studies, serum H2S levels negatively correlated with sputum neutrophil counts but positively correlated with sputum lymphocyte Citation[60] and macrophage Citation[62] counts, lung function (forced expiratory volume in 1 s) Citation[61,62] and serum •NO, suggesting that during COPD, H2S synthesis is induced in the lung to promote bronchial smooth muscle dilatation to improve airflow and limit inflammatory signaling.
In asthma, chronic inflammation of the airways leads to characteristic airway remodeling including subepithelial fibrosis and airway smooth muscle hypertrophy, resulting in irreversible narrowing of the airways (bronchoconstriction) Citation[63]. A role for H2S in asthma has also been investigated Citation[64]. In normal rat lung tissue, CSE expression is located primarily in airway and vascular smooth muscle cells Citation[64]. In the ovalbumin (OVA) challenge model of asthma in mice, OVA treatment reduced CSE expression and decreased pulmonary tissue H2S synthesis. Exogenous administration of NaSH reduced airway inflammation and decreased eosinophilic and neutrophilic influx. NaSH also attenuated OVA-induced pulmonary iNOS activation, reduced airway remodeling (goblet cell and smooth muscle cell hyperplasia and collagen deposition score) and restored peak expiratory flow (PEF). Furthermore, H2S concentrations in serum and lung positively correlated with PEF and negatively correlated with bronchoalveolar neutrophil and eosinophil count. These studies strongly suggest that induction of CSE-derived H2S during asthma plays an anti-inflammatory and antiremodeling role in the pathogenesis of asthma.
Although the precise molecular mechanisms by which H2S induces vascular remodeling and modulates inflammatory signaling in the lung are unknown, therapeutic modulation of endogenous H2S synthesis may be a viable approach for the treatment of chronic inflammatory lung diseases such as asthma and COPD.
H2S & inflammatory joint disease
Rheumatoid arthritis (RA) is the most common form of chronic inflammatory joint disease, affecting approximately 1% of the adult population in the Northern hemisphere Citation[65]. As such, RA is of considerable economic concern. RA is characterized by diffuse cartilage loss and the erosion of bone and cartilage. This process is believed to start in the synovial membrane with initial edema and hyperplasia of the synovial lining. Overproliferation of synoviocytes and macrophages causes thickening of the synovial lining, which together with lymphocytes, plasma cells and mast cells, develops into a sheet of invasive cellular tissue that is continuous with the synovial lining (pannus) Citation[4]. As a result of the higher proportion of synoviocytes, the pannus causes erosion of bone and cartilage at the margin of joints, resulting in the loss of cartilage-producing articular chondrocytes, joint erosion and eventual loss of joint mobility Citation[65,66].
Recently, we showed that synovial fluid (SF) aspirates from RA patients contained up to fourfold-higher concentrations of H2S than in paired plasma samples (RA SF median concentration: 62.41 µmol/l) and more than twofold-higher H2S levels than SF aspirates from patients with noninflammatory arthritides (median: 25.1 µmol/l) Citation[22]. Furthermore, SF H2S concentrations negatively correlated with SF neutrophil and total white cell counts, and positively correlated with tender joint counts, suggesting SF H2S could represent a novel index of disease activity in the inflamed joint. Increased synovial synthesis of H2S may not be specific to RA since other inflammatory joint diseases such as psoriatic and reactive arthritides also have significantly higher SF levels of H2S compared with matched plasma However, whether the increase in synovial H2S synthesis represents an endogenous mechanism to promote inflammation or whether synthesis was elevated in response to joint injury as an anti-inflammatory response is unknown. Recently, Kloesch et al. showed that in synviocytes isolated from RA patients, administration of a bolus of NaSH transiently decreased and then increased IL-1β-induced synthesis of IL-6 Citation[67]. Mechanistic studies suggested that this was independent of NF-κB activation but dependent on ERK1/2 deactivation. However, it should be noted that the predominant effects were seen at NaSH concentrations equal to or greater than 125 µmol/l (often 1 mmol/l), which are considerably higher than the reported levels of H2S in synovial fluid Citation[22]. Nevertheless, the same biphasic effect of H2S on inflammatory signaling has also been observed in LPS-treated murine macrophages where low concentrations of H2S inhibited LPS-induced synthesis of PGE2, •NO, IL-1β and IL-6 and NF-κB activity, but higher concentrations of NaSH promoted the synthesis of proinflammatory mediators Citation[68]. Furthermore, in an in vivo murine model of acute arthritis induced by kaolin/carrageenan Citation[69], a bolus addition of 30–50 µM of Na2S inhibited leukocyte adhesion in postcapillary venules in acutely inflamed mouse knees. In contrast to the effects of H2S on the systemic vasculature, Na2S induced vessel constriction rather than vasodilatation. Although the precise reasons for this observation are unclear, one possibility is that H2S induces vessel constriction to counteract local proinflammatory vasodilatory mediators such as PGE2, •NO and histamine. Together, these in vivo and in vitro studies would suggest that increased H2S synthesis in the inflamed human joint Citation[22] could represent a novel endogenous mechanism to control, limit or resolve inflammation, although further work is clearly required to substantiate this.
Role for H2S in neurogenic inflammation?
During neurogenic inflammation, the direct stimulation of afferent sensory nerves induces the release of bioactive inflammatory mediators, such as SP, NK-A and calcitonin gene-related peptide (CGRP). SP binding to NKR1 induces local vasodilatation, increased microvascular permeability and edema resulting in the accumulation of leukocytes. H2S has also been proposed as a novel mediator of neurogenic inflammation. Intraperitoneal injection of NaSH into ‘normal’ mice induced significant NKR1-dependent, but NKR2- and CGRP-independent, increases in plasma levels of SP and pronounced lung inflammation Citation[70], and in sepsis-induced lung injury Citation[71] and acute pancreatitis Citation[72], H2S upregulated plasma levels of SP and aggravated lung inflammation. These effects were blocked by either a selective NK1R antagonist or genetic depletion of PPT-A. In isolated murine pancreatic acinar cells, NaSH induced, but PAG inhibited, SP release and NK1R/PPT-A gene expression. Furthermore, in the isolated guinea pig airway, NaSH induced the release of SP and CGRP from sensory nerve terminals, and led to airway constriction through capsaicin and NKR1/NKR2-transient receptor potential vanilloid 1 (TRPV1)-dependent pathways Citation[73,74]. Similarly, in isolated rat bladder Citation[75] and rat detrusor muscle Citation[76], NaSH induced tachykin release from caspaicin-sensitive primary afferent neurons, leading to smooth muscle contraction. Collectively, these studies suggest that induction of neurogenic inflammation could mediate the reported proinflammatory role of H2S in inflammatory pathologies.
Antioxidant activity of H2S?
It has been long established that increased production of reactive oxygen species and at sites of acute and chronic inflammation is an important component of the inflammatory process Citation[4]. The resultant oxidative stress is an important component of the inflammatory process. For example, upregulation of •NO from iNOS, superoxide (O2•-) from endothelial cell and phagocytic cell NAD(P)H oxidase (respiratory burst), peroxynitrite (ONOO-) formed from the interaction of •NO with O2•-, hydrogen peroxide (H2O2) and hypochlorous acid (HOCl), derived from neutrophil MPO, are targeted towards the initial inflammatory insult, for example, bacterial infection. Overproduction of these reactive intermediates would not only damage the bacteria but would damage surrounding tissue, precipitating a further inflammatory response. Inhibition of ‘oxidative stress’ by inactivating enzymes or the direct ‘scavenging’ of the oxidant species would be expected to contribute to the many potential anti-inflammatory effects of H2S. In vitro experiments have shown that H2S generated from sulfide salts (Na2S and NaSH) inhibits cellular damage and intracellular protein oxidation induced by HOCl Citation[77,78], ONOO-Citation[79] and •NO Citation[34,80]. NaSH is also reported to scavenge and/or degrade lipid peroxides Citation[81,82] and inhibit the expression and activity of NAD(P)H oxidase Citation[83,84]. Increased hepatic glutathione (GSH) synthesis and decreased lipid peroxidation are also observed with Na2S treatment in a murine hepatic ischemia–reperfusion injury model Citation[85]. In neuronal cells, NaSH inhibited cell death induced by β-amyloid Citation[86] and MPP+Citation[87], mediated, at least in part, via antioxidant effects, and upregulated intracellular GSH synthesis through increasing cysteine uptake and elevation of glutathione-S-transferase activity Citation[88,89]. Furthermore, administration of Na2S to mice or overexpression of CSE increased Nrf-2 signaling Citation[90,91]. There is also emerging evidence for antioxidant activity in vivo in acute burn injury Citation[57] and myocardial injury Citation[90,91].
Are there ‘selective’ inhibitors of H2S synthesis?
A considerable amount of data on the potential role of endogenous H2S in inflammatory signaling has been obtained by the use of pharmacological inhibitors of CSE (e.g., DL-PAG and BCA) and inhibitors of CBS (e.g., AOAA) . While undoubtedly initially very useful pharmacological tools in a new but rapidly expanding research field, it is unlikely that these compounds are entirely specific since they target the pyridoxal phosphate (PLP) binding site of CSE and CBS Citation[92] and other PLP-dependent and -independent enzymes (summarized in ). This problem is exaggerated by the disparate concentration ranges commonly used in the laboratory (often used up to 10 mmol/l Citation[93–95]). Detailed pharmacokinetic analyses of these compounds have not been determined and the effective concentrations of these inhibitors are unknown. Although PAG, AOAA and BCA significantly reduce H2S synthesis in various animal models and the activity of CSE and CBS in isolated cells and tissues in vitro, over this concentration range, PAG and AOAA will exert significant effects independent of CSE or CBS inhibition. For example, in the rat colon, BCA induces submucosal edema and hypertrophy of the muscularis Citation[46], whereas PAG inhibits cardiac L-alanine transaminase activity Citation[96–98], and chronic dosing in rats led to increased heart, liver and spleen sizes Citation[99]. Similarly, AOAA has been widely used as a general inhibitor of transamination reactions Citation[97,98], inhibits the malate–aspartate shuttle, preventing lactate gluconeogenesis Citation[100,101], inhibits mitochondrial and cytosolic aspartate transaminase activity Citation[102,103] and inhibits protein synthesis Citation[104]. AOAA may exert at least some of its inhibitory effects on H2S metabolism through inhibition of 3-mercaptopyruvate sulfur transferase (3-MST) Citation[102,105], an additional enzymatic neuronal Citation[106] and vascular Citation[107] source of H2S. Although PAG rapidly accumulates in blood and through the D-amino acid oxidase pathway, it is metabolized to an as yet unidentified metabolite, leading to renal injury and significant proteinuria, glucosuria and polyuria Citation[108]. However, this was not observed with L-PAG as opposed to D-PAG. Since the kidney is the primary organ for fluid homeostasis, the renal and diuretic effects of this PAG metabolite (or PAG itself) are likely to contribute to (or confound) any experimental observations, in particular when assessing the precise role of CSE-derived H2S in swelling and tissue edema. These CSE-independent effects may offer at least one explanation for the disparate effects of H2S inhibitors and H2S donors in inflammation. Although AOAA, BCA and PAG clearly inhibit H2S synthesis in isolated tissue extracts, the true extent to which these compounds are able to penetrate cellular membranes and inhibit intracellular CSE and CBS is yet to be determined. It is possible that there is inconsistent cell penetration and cell metabolism among cell types and animal species, further contributing to the disparity in the literature. However, currently, there are no wholly specific CSE or CBS inhibitors available to researchers. Therefore, experimental data should be interpreted with the aforementioned observations in mind.
H2S donor molecules & inflammation: notes of caution
As with any new and emerging field of study, researchers are limited by the availability of specific, purpose-designed experimental tools. The vast majority of studies on H2S, in any area of physiology, as discussed previously, have utilized Na2S and NaSH as sources of H2S . While these salts are undoubtedly convenient and useful in that solutions of H2S (and HS-) can be readily prepared in the laboratory without the requirement for the use of awkward H2S gas cylinders, they are not particularly relevant tools to examine the physiology of H2S in vitro or in vivo. The addition of Na2S, NaSH or saturated solutions of H2S gas to aqueous solutions results in the instantaneous release of a bolus of H2S (and HS-), which dissipates in seconds Citation[33,68]. It is highly unlikely that cells or tissues are ever exposed to H2S generated in such a rapid manner, generating high local concentrations of H2S, since endogenously produced H2S through CSE and CBS is relatively slow and sustained Citation[68,92,109,110]. Furthermore, injection of these salts into animals results in a minimal increase in plasma H2S concentrations over a very short period of time Citation[21,33,68,111]. Commercially available NaSH rarely exceeds 70% purity and it is possible that the disparate physiological findings with NaSH could reflect the varying amounts of chemical impurities present in the NaSH. Commercial Na2S, on the other hand, is considerably purer but in in vitro experiments, still generates H2S too quickly to be physiologically equivalent. Nonetheless, as a therapeutic tool, pharmaceutical-grade Na2S shows great potential and has been successfully used to prevent and treat myocardial infarction, ischemia–reperfusion injury and endotoxic shock Citation[112,113]. It is unclear whether these effects are directly due to released H2S or whether H2S is sequestered into biologically active intermediaries (e.g., of plasma protein or lipids; also reviewed in detail elsewhere in this issue), further highlighting the complex nature of H2S physiology and medicine.
Future developments: slow-release H2S donor molecules
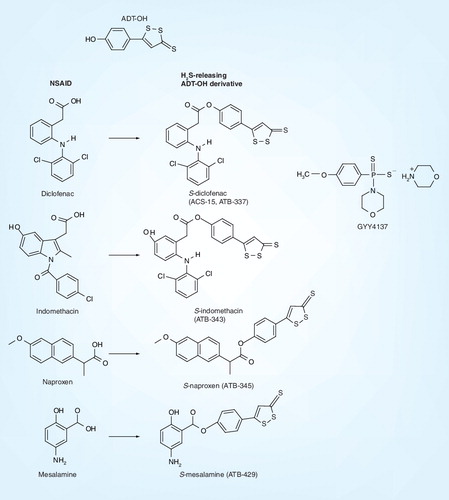
The problem of using rapidly generated H2S from a bolus of concentrated sulfide salt is currently being addressed. Recently H2S donors have been synthesized to circumvent this problem using 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione (ADT-OH), derivatives of existing NSAID molecules, such as indomethacin (ATB-343), diclofenac (ACS-15), naproxen (ATB-345) and mesalamine (ATB-429), reviewed in detail elsewhere in this issue. These derivatives have shown great promise in alleviating and limiting gastrointestinal side effects and toxicity of NSAIDs and in the treatment of experimental inflammatory bowel disease and arthritis (summarized in ).
However, it should be noted that ADT-OH is the major metabolite of 3H-1,2-dithiole-3-thione (ADT; a dithiolethione), a putative chemopreventative agent structurally related to olitipraz. Dithiolethiones and related compounds have previously been explored as inhibitors of carcinogenesis and have shown considerable promise in animal models of edema and gastrointestinal inflammation (extensively reviewed in Citation[114,115]; summarized in ). However, it is possible that some of the observed biological effects associated with ADT-OH derivatives were due to the dithiolethione moiety itself, rather than released H2S. Indeed, an examination of the pertinent literature shows that dithiolethiones, including ADT-OH, are reactive with thiols and elicit several effects that may not be due to H2S. For example, Isenberg et al.Citation[116] and Moody et al.Citation[117] recently showed that the dithiolethione moiety of ADT-OH-modified NSAIDs and valproate rather than released H2S inhibited angiogenesis and cell proliferation. Others have shown dithiolethiones to induce the synthesis of phase II enzymes (e.g., glutathione-S-transferase, NAD[P]H, quinone oxidoreductase), GSH reductase and catalase Citation[118–122]. Similar to ADT, ADT-OH itself is also expected to elicit antioxidant activity in vivo. The dithiolethione derivatives are convenient, promising in animal studies of pathology as novel therapeutic agents, and represent a considerable advance in studying the physiology, pharmacology and pathology of H2S. However, care should therefore be taken to interpret experimental data with the above observations in mind. Control experiments consisting of the dithiolethione alone, parent compound, parent compound with ADT-OH and ‘decomposed’ donor molecule should be thoroughly evaluated and the release of H2S under each experimental condition should be measured.
More recently, a H2S donor that does not consist of a structurally modified NSAID molecule, GYY4137 Citation[32,33,68], has been synthesized and characterized (summarized in ). GYY4137 is a very slow-releasing H2S donor compound that releases two molecules of H2S per molecule of GYY4137 and has been shown to exert prominent endothelium-dependent vasodilatory activity in vivo via KATP channel-dependent mechanisms in hypertensive rats Citation[33]. GYY4137 also exerts prominent anti-inflammatory activity in endotoxic shock Citation[32] and in isolated inflammatory cells Citation[68], mediated in part via inhibition of NF-κB/AP-1-dependent proinflammatory signaling (summarized in ). In contrast to ADT-OH derivatives, GYY4137 is freely soluble in water and, unlike S-diclofenac Citation[123], GYY4137 is apparently not toxic to vascular cells or anesthetized rats, even when tested at mmol/l concentrations Citation[32,33]. GYY4137 offers an additional advantage to the researcher over ADT-OH compounds in that its decomposition products appear inactive Citation[68] allowing one to study the physiological effects of slow release of H2S in inflammation directly, in the absence of any possible effects of NSAID or ADT-OH. The precise metabolic profile for GYY4137 has yet to be elucidated so it is possible that at least some of the reported effects in vivo could be due to metabolism to biologically active intermediates.
Expert commentary & five-year view: is H2S pro- or anti-inflammatory?
Enzyme inhibitors, such as PAG, AOAA and BCA and sulfide salt H2S donors (e.g., Na2S and NaSH), have been useful in highlighting the potential role of a previously unrecognized endogenous biologically active gas as a novel mediator of inflammatory signaling. The bulk of the studies that have used either Na2S or NaSH as the source of H2S, or have used PAG (and BCA) and AOAA to inhibit CSE and CBS activity, have generally come to the conclusion that endogenous H2S is proinflammatory (summarized in ). However, Na2S and NaSH generate H2S too quickly to be an effective model for endogenously synthesized H2S. While PAG, BCA and AOAA decrease blood and tissue levels of H2S, they are not wholly specific, detailed pharmacodynamic and pharmacokinetic profiles have yet to be determined and the ‘effective’ concentration of drug in plasma and/or tissues is not known. Potential effects of these inhibitors, independent of H2S synthesis, especially in vivo, also cannot be ruled out. In order for the field to progress, pharmacological tools to selectively inactivate or inhibit CSE and CBS are required. Genetic approaches to remove CSE have made significant progress in unraveling the role of H2S in regulating blood pressure Citation[13] – although this is also an area of controversy Citation[124] – and these genetic tools have yet to be applied to inflammatory models. Similarly, overexpression of CSE and/or CBS in animals will also shed light on the role of endogenous H2S in regulating, driving or inhibiting inflammation.
Newer approaches to study H2S using ADT-OH derivatives or GYY4137 have thus far exclusively shown in a number of cellular and in vivo animal models that slow release of H2S, which more closely models H2S derived from CSE or CBS, is anti-inflammatory. This suggests that the observed effect of H2S in an experimental system may be dependent upon the manner in which cells and tissues are exposed to H2S. However, even slow-release H2S donors represent pharmacological H2S. By analogy to the •NO research field, it appears that we are still in the early stages of understanding the true significance of H2S in the inflammatory response.
In conclusion, the precise role of H2S in the inflammatory process is ambiguous (summarized in ). There is a great deal of disparity in the literature concerning the anti-inflammatory effects (‘the good’), and the proinflammatory effects (‘the bad’) of endogenous and pharmacological H2S. As with any new and emerging field, researchers are limited by the availability of suitable experimental tools. In particular, progress has been hampered by a lack of availability of enzyme-specific inhibitors and physiologically irrelevant H2S donors, which have often been used at very high concentrations (‘the ugly’). The field is further impeded by the current lack of detailed pharmacodynamic and pharmocokinetic profiling of donors and inhibitors. In sharp contrast to the use of sulfide salts (e.g., Na2S and NaSH) as a source of pharmacological H2S, the development of novel donors that can deliver H2S in a slow and sustained manner to model enzymatically synthesized H2S has universally shown anti-inflammatory activity (‘the promising’). These new molecules offer the potential to clarify the precise role of H2S in acute and chronic inflammation, as well as to provide an additional class of therapeutic compounds for other emerging aspects of H2S physiology.
Table 1. Examples of the reported effects of commonly used inhibitors of H2S synthesis in vitro and in vivo, highlighting the nonspecific nature of these compounds.
Table 2. Evidence consistent with an anti-inflammatory role of endogenous and exogenous (pharmacological) H2S.
Table 3. Evidence consistent with a proinflammatory role of endogenous and exogenous (pharmacological) H2S.
Table 4. Summary of the effects of exogenous and endogenous H2S on inflammation and edema.
Key issues
• The pro- and anti-inflammatory actions of hydrogen sulfide (H2S) involve multiple pathways and are complex.
• The observed pro- and anti-inflammatory effects of H2S may be influenced by the exact nature of the model of inflammation (e.g., acute vs chronic inflammation), and the timing of the administration of a H2S donor or inhibiting compound.
• The effects of H2S in models of inflammation are concentration and time dependent, and may involve ‘bell-shaped’ dose–response curves.
• Slow-release H2S donors are a more physiologically relevant source of H2S than sulfide salts.
• Commonly used inhibitors of cystathionine γ-lyase and cystathionine β-synthase are not wholly specific, and careful control experiments are required to rule-out H2S-independent effects.
Financial & competing interests disclosure
The authors would like to thank the Wellcome Trust (#091725), the European Union FP7 (RedCat #215009) and the Devon Arthritis Appeal Research Trust for continued financial support. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy4, 281–286 (2005).
- Pathological Basis of Disease. Ramzi VK, Contran S, Tucker C (Eds). W Saunders B, NY, USA 1999.
- What Is Inflammation? Winyard PG, Blake DR, Evans CH (Eds). Birkhauser Verlag, Basel, Swizerland (2000).
- Pattison DJ, Winyard PG. Dietary antioxidants in inflammatory arthritis: do they have any role in etiology or therapy? Nat. Clin. Pract. Rheumatol.4, 590–596 (2008).
- Key Stages in the Acute Inflammatory Response and Their Relevance as Therapeutic Targets. Winyard PG, Willoughby D (Eds). Humana Press, NJ, USA (2003).
- Li L, Hsu A, Moore PK. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation – a tale of three gases! Pharmacol. Ther.123, 386–400 (2009).
- Pae HO, Lee YC, Jo EK, Chung HT. Subtle interplay of endogenous bioactive gases (NO, CO and H2S) in inflammation. Arch. Pharm. Res.32, 1155–1162 (2009).
- Kaczorowski DJ, Zuckerbraun BS. Carbon monoxide: medicinal chemistry and biological effects. Curr. Med. Chem.14, 2720–2725 (2007).
- Nitric Oxide and Inflammation. Salvemini D, Billiar TM, Vodovotz Y (Eds). Birkhauser Verlag, Basel, Swizerland (2001).
- Laskin JD, Heck DE, Laskin DL. Multifunctional role of nitric oxide in inflammation. Trends Endocrinol. Metab.5, 377–382 (1994).
- Li L, Bhatia M, Moore PK. Hydrogen sulphide – a novel mediator of inflammation? Curr. Opin. Pharmacol.6, 125–129 (2006).
- Whiteman M, Moore PK. Hydrogen sulfide and the vasculature: a novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell. Mol. Med.13, 488–507 (2009).
- Yang G, Wu L, Jiang B et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science322, 587–590 (2008).
- Ali MY, Whiteman M, Low CM, Moore PK. Hydrogen sulphide reduces insulin secretion from HIT-T15 cells by a KATP channel-dependent pathway. J. Endocrinol.195, 105–112 (2007).
- Yang W, Yang G, Jia X, Wu L, Wang R. Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J. Physiol.569, 519–531 (2005).
- Smith HS. Hydrogen sulfide’s involvement in modulating nociception. Pain Physician12, 901–910 (2009).
- Kimura H. Hydrogen sulfide as a neuromodulator. Mol. Neurobiol.26, 13–19 (2002).
- Whiteman M, Gooding KM, Whatmore JL et al. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia53, 1722–1726 (2010).
- Jin HF, Liang C, Liang JM, Tang CS, Du JB. Effects of hydrogen sulfide on vascular inflammation in pulmonary hypertension induced by high pulmonary blood flow: experiment with rats. Zhonghua Yi Xue Za Zhi88, 2235–2239 (2008).
- Mok YY, Atan MS, Ping CY et al. Role of hydrogen sulphide in haemorrhagic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. Br. J. Pharmacol.143, 881–889 (2004).
- Li L, Bhatia M, Zhu YZ et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J.19, 1196–1198 (2005).
- Whiteman M, Haigh R, Tarr JM, Gooding KM, Shore AC, Winyard PG. Detection of hydrogen sulfide in plasma and knee-joint synovial fluid from rheumatoid arthritis patients: relation to clinical and laboratory measures of inflammation. Ann. NY Acad. Sci.1203, 146–150 (2010).
- Martin GR, McKnight GW, Dicay MS, Coffin CS, Ferraz JG, Wallace JL. Hydrogen sulphide synthesis in the rat and mouse gastrointestinal tract. Dig. Liver Dis.42, 103–109 (2010).
- Astiz ME, Rackow EC. Septic shock. Lancet351, 1501–1505 (1998).
- Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA273, 117–123 (1995).
- Gardiner SM, Kemp PA, March JE, Bennett T. Regional haemodynamic responses to infusion of lipopolysaccharide in conscious rats: effects of pre- or post-treatment with glibenclamide. Br. J. Pharmacol.128, 1772–1778 (1999).
- Shi W, Cui N, Wu Z et al. Lipopolysaccharides up-regulate Kir6.1/SUR2B channel expression and enhance vascular KATP channel activity via NF-κB-dependent signaling. J. Biol. Chem.285, 3021–3029 (2010).
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J.20, 6008–6016 (2001).
- Hui Y, Du J, Tang C, Bin G, Jiang H. Changes in arterial hydrogen sulfide (H2S) content during septic shock and endotoxin shock in rats. J. Infect.47, 155–160 (2003).
- Zhang H, Zhi L, Moore PK, Bhatia M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol.290, L1193–L1201 (2006).
- Collin M, Anuar FB, Murch O, Bhatia M, Moore PK, Thiemermann C. Inhibition of endogenous hydrogen sulfide formation reduces the organ injury caused by endotoxemia. Br. J. Pharmacol.146, 498–505 (2005).
- Li L, Salto-Tellez M, Tan CH, Whiteman M, Moore PK. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic. Biol. Med.47, 103–113 (2009).
- Li L, Whiteman M, Guan YY et al. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation117, 2351–2360 (2008).
- Ali MY, Ping CY, Mok YY et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol.149, 625–634 (2006).
- Mok YY, Moore PK. Hydrogen sulphide is pro-inflammatory in haemorrhagic shock. Inflamm. Res.57, 512–518 (2008).
- Zhang H, Moochhala SM, Bhatia M. Endogenous hydrogen sulfide regulates inflammatory response by activating the ERK pathway in polymicrobial sepsis. J. Immunol.181, 4320–4331 (2008).
- Zhang H, Zhi L, Moochhala S, Moore PK, Bhatia M. Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-κB. Am. J. Physiol. Lung Cell. Mol. Physiol.292, L960–L971 (2007).
- Bhatia M, Zhi L, Zhang H, Ng SW, Moore PK. Role of substance P in hydrogen sulfide-induced pulmonary inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol.291, L896–L904 (2006).
- Inflammation Protocols: Carrageenan-Induced Paw Edema in the Rat and Mouse. Morris CJ (Ed.). Humana Press, NJ, USA (2003).
- Bhatia M, Sidhapuriwala J, Moochhala SM, Moore PK. Hydrogen sulphide is a mediator of carrageenan-induced hindpaw oedema in the rat. Br. J. Pharmacol.145, 141–144 (2005).
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J.20, 2118–2120 (2006).
- Wallace JL, Granger DN. The cellular and molecular basis of gastric mucosal defense. FASEB J.10, 731–740 (1996).
- Wallace JL. Prostaglandins. NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol. Rev.88, 1547–1565 (2008).
- Sigthorsson G, Simpson RJ, Walley M et al. COX-1 and 2, intestinal integrity, and pathogenesis of nonsteroidal anti-inflammatory drug enteropathy in mice. Gastroenterology122, 1913–1923 (2002).
- Fiorucci S, Antonelli E, Distrutti E et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology129, 1210–1224 (2005).
- Wallace JL, Vong L, W McKnight. Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology137, 569–578, 578 e561 (2009).
- Wallace JL, Dicay M, W McKnight. Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J.21, 4070–4076 (2007).
- Abeles RH, Walsh CT. Acetylenic enzyme inactivators. Inactivation of g-cystathionase, in vitro and in vivo, by propargylglycine. J. Am. Chem. Soc.95, 6124–6125 (1973).
- Pfeffer M, Ressler C. β-cyanoalanine, an inhibitor of rat liver cystathionase. Biochem. Pharmacol.16, 2299–2308 (1967).
- Tamizhselvi R, Moore PK, Bhatia M. Hydrogen sulfide acts as a mediator of inflammation in acute pancreatitis: in vitro studies using isolated mouse pancreatic acinar cells. J. Cell. Mol. Med.11, 315–326 (2007).
- Tamizhselvi R, Moore PK, Bhatia M. Inhibition of hydrogen sulfide synthesis attenuates chemokine production and protects mice against acute pancreatitis and associated lung injury. Pancreas36, e24–e31 (2008).
- Tamizhselvi R, Koh YH, Sun J, Zhang H, Bhatia M. Hydrogen sulfide induces ICAM-1 expression and neutrophil adhesion to cerulein-treated pancreatic acinar cells through NF-κB and Src-family kinases pathway. Exp. Cell. Res.316, 1625–1636 (2010).
- Adhikari S, Bhatia M. H2S-induced pancreatic acinar cell apoptosis is mediated via JNK and p38 MAP kinase. J. Cell. Mol. Med.12, 1374–1383 (2008).
- Cao Y, Adhikari S, Ang AD, Moore PK, Bhatia M. Mechanism of induction of pancreatic acinar cell apoptosis by hydrogen sulfide. Am. J. Physiol. Cell. Physiol.291, C503–C510 (2006).
- Sidhapuriwala JN, Ng SW, Bhatia M. Effects of hydrogen sulfide on inflammation in cerulein-induced acute pancreatitis. J. Inflamm. (Lond.)6, 35 (2009).
- Zhang J, Sio SW, Moochhala S, Bhatia M. Role of hydrogen sulfide in severe burn injury-induced inflammation in mice. Mol. Med.16, 417–424 (2010).
- Esechie A, Kiss L, Olah G et al. Protective effect of hydrogen sulfide in a murine model of acute lung injury induced by combined burn and smoke inhalation. Clin. Sci. (Lond.)115, 91–97 (2008).
- MD Barber RC, White DJ, Horton JW. Increasing percent burn is correlated with increasing inflammation in an adult rodent model. Shock30, 388–393 (2008).
- Lopez A, Prior MG, Reiffenstein RJ, Goodwin LR. Peracute toxic effects of inhaled hydrogen sulfide and injected sodium hydrosulfide on the lungs of rats. Fundam. Appl. Toxicol.12, 367–373 (1989).
- Lopez A, Prior M, Lillie LE, Gulayets C, Atwal OS. Histologic and ultrastructural alterations in lungs of rats exposed to sub-lethal concentrations of hydrogen sulfide. Vet. Pathol.25, 376–384 (1988).
- Chen YH, Yao WZ, Geng B et al. Endogenous hydrogen sulfide in patients with COPD. Chest128, 3205–3211 (2005).
- Chen YH, Yao WZ, Ding YL, Geng B, Lu M, Tang CS. Effect of theophylline on endogenous hydrogen sulfide production in patients with COPD. Pulm. Pharmacol. Ther.21, 40–46 (2008).
- Bateman ED, Hurd SS, Barnes PJ et al. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J.31, 143–178 (2008).
- Chen YH, Wu R, Geng B et al. Endogenous hydrogen sulfide reduces airway inflammation and remodeling in a rat model of asthma. Cytokine45, 117–123 (2009).
- Symmons D, Turner G, Webb R et al. The prevalence of rheumatoid arthritis in the United Kingdom: new estimates for a new century. Rheumatology (Oxford)41, 793–800 (2002).
- Scott DL, Willoughby DA, Blake DR. Molecular insights into rheumatoid arthritis. Mol. Aspects Med.12, 341–394 (1991).
- Kloesch B, Liszt M, Broell J. H2S transiently blocks IL-6 expression in rheumatoid arthritic fibroblast-like synoviocytes and deactivates p44/42 mitogen-activated protein kinase. Cell. Biol. Int.34, 477–484 (2010).
- Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox Signal.12, 1147–1154 (2010).
- Andruski B, McCafferty DM, Ignacy T, Millen B, McDougall JJ. Leukocyte trafficking and pain behavioral responses to a hydrogen sulfide donor in acute monoarthritis. Am. J. Physiol. Regul. Integr. Comp. Physiol.295, R814–R820 (2008).
- Bhatia M, Wong FL, Fu D, Lau HY, Moochhala SM, Moore PK. Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J.19, 623–625 (2005).
- Zhang H, Hegde A, Ng SW, Adhikari S, Moochhala SM, Bhatia M. Hydrogen sulfide up-regulates substance P in polymicrobial sepsis-associated lung injury. J. Immunol.179, 4153–4160 (2007).
- Zhang H, Zhi L, Moochhala SM, Moore PK, Bhatia M. Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J. Leukoc. Biol.82, 894–905 (2007).
- Trevisani M, Patacchini R, Nicoletti P et al. Hydrogen sulfide causes vanilloid receptor 1-mediated neurogenic inflammation in the airways. Br. J. Pharmacol.145, 1123–1131 (2005).
- Ang SF, Moochhala SM, Bhatia M. Hydrogen sulfidde promotes transient receptor potential vanilloid 1-mediated neurogenic inflammation in polymicrobial sepsis. Crit. Care Med.38, 619–628 (2010).
- Patacchini R, Santicioli P, Giuliani S, Maggi CA. Pharmacological investigation of hydrogen sulfide (H2S) contractile activity in rat detrusor muscle. Eur. J. Pharmacol.509, 171–177 (2005).
- Patacchini R, Santicioli P, Giuliani S, Maggi CA. Hydrogen sulfide (H2S) stimulates capsaicin-sensitive primary afferent neurons in the rat urinary bladder. Br. J. Pharmacol.142, 31–34 (2004).
- Whiteman M, Cheung NS, Zhu YZ et al. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun.326, 794–798 (2005).
- Laggner H, Muellner MK, Schreier S et al. Hydrogen sulphide: a novel physiological inhibitor of LDL atherogenic modification by HOCl. Free Radic. Res.41, 741–747 (2007).
- Whiteman M, Armstrong JS, Chu SH et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J. Neurochem.90, 765–768 (2004).
- Whiteman M, Li L, Kostetski I et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun.343, 303–310 (2006).
- Muellner MK, Schreier SM, Laggner H et al. Hydrogen sulfide destroys lipid hydroperoxides in oxidized LDL. Biochem. J.420, 277–281 (2009).
- Schreier SM, Muellner MK, Steinkellner H et al. Hydrogen sulfide scavenges the cytotoxic lipid oxidation product 4-HNE. Neurotox. Res.17, 249–256 (2010).
- Tyagi N, Moshal KS, Sen U et al. H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid. Redox Signal.11, 25–33 (2009).
- Muzaffar S, Shukla N, Bond M et al. Exogenous hydrogen sulfide inhibits superoxide formation. NOX-1 expression and Rac1 activity in human vascular smooth muscle cells. J. Vasc. Res.45, 521–528 (2008).
- Jha S, Calvert JW, Duranski MR, Ramachandran A. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signalling. Am. J. Physiol. Heart Circ. Physiol.295, H801–H806 (2008).
- Liu YY, Bian JS. Hydrogen sulfide protects amyloid-β induced cell toxicity in microglia. J. Alzheimers Dis. (2010) (Epub ahead of print).
- Yin WL, He JQ, Hu B, Jiang ZS, Tang XQ. Hydrogen sulfide inhibits MPP(+)-induced apoptosis in PC12 cells. Life Sci.85, 269–275 (2009).
- Kimura Y, Dargusch R, Schubert D, Kimura H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid. Redox Signal.8, 661–670 (2006).
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J.18, 1165–1167 (2004).
- Calvert JW, Jha S, Gundewar S et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res.105, 365–374 (2009).
- Calvert JW, Elston M, Nicholson CK et al. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation122, 11–19 (2010).
- Sun Q, Collins R, Huang S et al. Structural basis for the inhibition mechanism of human cystathionine γ-lyase, an enzyme responsible for the production of H2S. J. Biol. Chem.284, 3076–3085 (2009).
- d’Emmanuele R, di Villa B, Sorrentino R et al. Hydrogen sulfide as a mediator of human corpus cavernosum smooth-muscle relaxation. Proc. Natl Acad. Sci. USA106, 4513–4518 (2009).
- Stecyk JA, Skovgaard N, Nilsson GE, Wang T. Vasoactivity of hydrogen sulfide in normoxic and anoxic turtles (Trachemys scripta). Am. J. Physiol. Regul. Integr. Comp. Physiol.298, R1225–R1239 (2010).
- Bucci M, Mirone V, Di Lorenzo A et al. Hydrogen sulphide is involved in testosterone vascular effect. Eur. Urol.56, 378–383 (2009).
- Burnett G, Marcotte P, Walsh C. Mechanism-based inactivation of pig heart L-alanine transaminase by L-propargylglycine. Half-site reactivity. J. Biol. Chem.255, 3487–3491 (1980).
- Jerebzoff S. Are there ultraradian rhythms at the molecular level? Biol. Rhythm Res.18, 9–16 (1997).
- Subramanian RK, Kasumov T, Yang L et al. Reassessment of the mechanisms by which aminooxyacetate (AOA) inhibits gluconeogenesis (GNG) from lactate. FASEB J.21, 804–811 (2007).
- Cho ES, Hovanec-Brown J, Tomanek RJ, Stegink LD. Propargylglycine infusion effects on tissue glutathione levels, plasma amino acid concentrations and tissue morphology in parenterally-fed growing rats. J. Nutr.121, 785–794 (1991).
- Ochs RS, Harris RA. Aminooxyacetate inhibits gluconeogenesis by isolated chicken hepatocytes. Biochim. Biophys. Acta632, 260–269 (1980).
- Stottrup NB, Lofgren B, Birkler RD et al. Inhibition of the malate–aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc. Res.88(2), 257–266 (2010)
- Ubuku AT, Fujiwara M, Wrobel M. Inhibition of sulfate excretion by (aminooxy)acetate induced stimulation of taurine excretion in rats. Amino Acids8, 345–352 (2004).
- Reg R. Aminooxyacetate is not an adequate differential inhibitor of aspartate aminotransferase isoenzymes. Clin. Chem.23, 1508–1509 (1977).
- Girbes T, Alonso P. Inhibition of protein synthesis by (aminooxy)acetate in rat liver. Int. J. Biochem.8, 537–542 (1986).
- Teraoka T, Ohta J, Abe T, Inoue H. Inhibition of sulfate-forming activity in rat liver mitochondria by (aminooxy)acetate. Amino Acids5, 245–251 (2004).
- Laggner H, Hermann M, Sturm B, Gmeiner BM, Kapiotis S. Sulfite facilitates LDL lipid oxidation by transition metal ions: a pro-oxidant in wine? FEBS Lett.579, 6486–6492 (2005).
- Kapiotis S, Hermann M, Exner M, Laggner H, Gmeiner BM. Copper- and magnesium protoporphyrin complexes inhibit oxidative modification of LDL induced by hemin, transition metal ions and tyrosyl radicals. Free Radic. Res.39, 1193–1202 (2005).
- Konno R, Ikeda M, Yamaguchi K, Ueda Y, Niwa A. Nephrotoxicity of d-propargylglycine in mice. Arch. Toxicol.74, 473–479 (2000).
- Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, Banerjee R. H2S biogenesis by human cystathionine γ-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem.284, 11601–11612 (2009).
- Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine β-synthase and g-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem.284, 22457–22466 (2009).
- Fiorucci S, Orlandi S, Mencarelli A et al. Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br. J. Pharmacol.150, 996–1002 (2007).
- Sodha NR, Clements RT, Feng J et al. The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia–reperfusion injury. Eur. J. Cardiothorac. Surg.33, 906–913 (2008).
- Simon F, Giudici R, Duy CN et al. Hemodynamic and metabolic effects of hydrogen sulfide during porcine ischemia/reperfusion injury. Shock30, 359–364 (2008).
- Wallace JL. Building a better aspirin: gaseous solutions to a century-old problem. Br. J. Pharmacol.152, 421–428 (2007).
- Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol. Sci.28, 501–505 (2007).
- Isenberg JS, Jia Y, Field L et al. Modulation of angiogenesis by dithiolethione-modified NSAIDs and valproic acid. Br. J. Pharmacol.151, 63–72 (2007).
- Moody TW, Switzer C, Santana-Flores W et al. Dithiolethione modified valproate and diclofenac increase E-cadherin expression and decrease proliferation of non-small cell lung cancer cells. Lung Cancer68, 154–160 (2010).
- Munday R, Munday CM. Selective induction of Phase II enzymes in the urinary bladder of rats by allyl isothiocyanate, a compound derived from Brassica vegetables. Nutr. Cancer44, 52–59 (2002).
- Munday R, Munday CM. Induction of phase II enzymes by 3H-1,2-dithiole-3-thione: dose–response study in rats. Carcinogenesis25, 1721–1725 (2004).
- Munday R, Zhang Y, Munday CM, Li J. Structure-activity relationships in the induction of Phase II enzymes by derivatives of 3H-1,2-dithiole-3-thione in rats. Chem. Biol. Interact.160, 115–122 (2006).
- Kwak MK, Egner PA, Dolan PM et al. Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat. Res.480, 305–315 (2001).
- Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1–Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem.278, 8135–8145 (2003).
- Baskar R, Sparatore A, Del Soldato P. Moore PK. Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur. J. Pharmacol.594, 1–8 (2008).
- Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine γ-lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem.285, 26358–26368 (2010).
- Bhatia M, Sidhapuriwala JN, Ng SW, Tamizhselvi R, Moochhala SM. Pro-inflammatory effects of hydrogen sulphide on substance P in cerulein-induced acute pancreatitis. J. Cell. Mol. Med.12, 580–590 (2008).
- Zhi L, Ang AD, Zhang H, Moore PK, Bhatia M. Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK–NF-κB pathway. J. Leukoc. Biol.81, 1322–1332 (2007).
- Lee AT, Shah JJ, Li L, Cheng Y, Moore PK, Khanna S. A nociceptive-intensity-dependent role for hydrogen sulphide in the formalin model of persistent inflammatory pain. Neuroscience152, 89–96 (2008).
- Cunha TM, Dal-Secco D, Verri WA Jr et al. Dual role of hydrogen sulfide in mechanical inflammatory hypernociception. Eur. J. Pharmacol.590, 127–135 (2008).
- Bhatia M, Sidhapuriwala JN, Sparatore A, Moore PK. Treatment with H2S-releasing diclofenac protects mice against acute pancreatitis-associated lung injury. Shock29, 84–88 (2008).
- Li T, Zhao B, Wang C et al. Regulatory effects of hydrogen sulfide on IL-6, IL-8 and IL-10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp. Biol. Med. (Maywood)233, 1081–1087 (2008).
- Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic. Biol. Med.42, 706–719 (2007).
- Mariggio MA, Pettini F, Fumarulo R. Sulfide influence on polymorphonuclear functions: a possible role for Ca2+ involvement. Immunopharmacol. Immunotoxicol.19, 393–404 (1997).
- Persson S, Claesson R, Carlsson J. Chemotaxis and degranulation of polymorphonuclear leukocytes in the presence of sulfide. Oral Microbiol. Immunol.8, 46–49 (1993).
- Sidhapuriwala J, Li L, Sparatore A, Bhatia M, Moore PK. Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative, on carrageenan-induced hindpaw oedema formation in the rat. Eur. J. Pharmacol.569, 149–154 (2007).
- Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br. J. Pharmacol.159, 1236–1246 (2010).
- Cho ES, Havanec-Brown J, Tomanek RJ. Propargylglycine infusion effects on tissue glutathione levels, plasma amino acid concentrations and tissue morphology in parentally-fed growing rats. J. Nutr.121, 785–794 (1991).
- Jainyang Z, Machida Y, Sugahara K. Determination of D,L-propargyglycine and N-acetylpropargylglycine in urine and several tissues of D,L-propargylglycine-treated rats using liquid chromatography mass spectrometry. J. Chromatog. B660 (1994).
- Stottrup NB, Lofgren B, Birkler RD et al. Inhibition of the malate–aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc. Res.88, 257–266 (2010).
- Ubuku T, Abe T, Fujiwara M, Wrobe M. Inhibition of sulfate excretion by (aminooxy)acetate induced stimulation of taurine excretion in rats. Amino Acids8, 345–352 (2004).
- Malaisse WJ, Malaisse-Lagae F, Sener A. The stimulus-secretion coupling of glucose-induced insulin release: effect of aminooxyacetate upon nutrient-stimulated insulin secretion. Endocrinology111, 392–397 (1982).
- Gao Z, Young RA, Li G et al. Distinguishing features of leucine and α-ketoisocaproate sensing in pancreatic β-cells. Endocrinology144, 1949–1957 (2003).
- Heissig H, Urban KA, Hastedt K, Zunkler BJ, Panten U. Mechanism of the insulin-releasing action of α-ketoisocaproate and related α-keto acid anions. Mol. Pharmacol.68, 1097–1105 (2005).
- Arnett FC, Edworthy SM, Bloch DA et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum.31, 315–324 (1988).