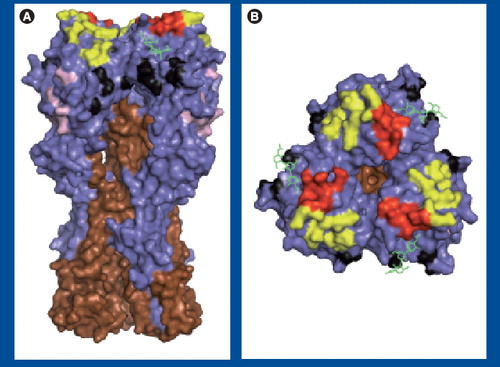
(A) Side and (B) top views of A/PR/8/34 HA (Protein Database: 1RVZ). HA1 and HA2 are shown in blue and brown, respectively, and sialic acid is shown in green. Antigenic sites are shown: Sa (yellow), Sb (red), Ca (black) and Cb (pink). Images were generated using Pymol software (DeLano Scientific LLC, CA, USA).
A new human influenza A virus (IAV) strain of swine-like origin emerged in Mexico and the USA in March and April of 2009 Citation[1]. The virus disseminated globally with such celerity that on 11 June 2009, the WHO raised the Flu Pandemic Alert Level to Phase 6, officially designating a new global pandemic. At the time, there were 30,000 confirmed cases in 74 countries. Less than 1 month later (as of 1 July 2009), the WHO confirmed over 77,000 cases and 332 deaths worldwide.
The collaborative nature of the global response to the outbreak has been remarkable. Within days of the initial reporting of the virus, researchers completely sequenced the genome of several viral isolates and posted the sequences on the internet. Sequences of new isolates continue to be uploaded daily on websites such as the Global Initiative on Sharing Avian Influenza Data Citation[101] and the National Center for Biotechnology Information Citation[102]. Nearly in real time, phylogeneticists identified the complex origins of the virus. Within weeks, a vaccine seed stock was isolated, and the private and public sector began intense collaborative efforts to produce a vaccine post-haste Citation[2].
This is the first major introduction of a pandemic influenza strain since 1977, and the first to benefit from the biotechnology revolution. Those looking to defend the investment of world governments into biomedical research have another shining example of the enormous payoff in both lives saved and money gained from achieving a better understanding of scientific processes. Retrospectively, concerns over the pandemic potential of avian IAVs prepared society well for the current pandemic, as recently upgraded resources have played critical roles in monitoring and responding rapidly. Scientists, physicians, administrators and politicians who fought for these resources deserve the gratitude of their nations.
Clearly, appropriate actions have been taken to limit disease and death associated with the new virus. The contrast with the 1918 pandemic, whose causative agent was not known for 13 years, could not be starker. Despite our advances, it is critical to recognize that today, as in 1918, humanity can neither control nor predict the genetic and antigenic evolution of this virus. Here we discuss factors in the evolution of 2009 swine-origin H1N1 IAV (hereafter referred to as S-IAV-2009).
Overview of IAV proteins
Influenza A viruses are negative ssRNA viruses with eight gene segments encoding at least 11 functional proteins. Virions consist of a matrix (M1) shell covered by a lipid membrane. Within the shell, the eight segments are coated with nucleoprotein (NP) and small quantities of the viral polymerase complex (PB1, PB2 and PA), and NS2 (also known as nuclear export protein). Two viral glycoproteins are embedded in the membrane: hemagglutinin (HA) and neuraminidase (NA). HA mediates binding to cellular receptors, which are sialic acid moieties terminally linked to oligosaccharides on cellular proteins and lipids. NA cleaves sialic acid, playing a critical role in progeny virus release from host cells and in preventing viral aggregation. Another small viral nonglycosylated membrane protein, M2, is an ion channel involved in viral entry and exit. The virus encodes two proteins excluded from virions: NS1 and PB1-F2. NS1 blocks innate immune antiviral responses and contributes to viral gene expression. While the functions of PB1-F2 remain to be firmly established, it appears to play an important role in pathogenicity Citation[3]. PB1-F2 is encoded by an overlapping reading frame in the PB1 gene, and contemporary H1 subtype viruses express a truncated form of less than 60 amino acids (compared with ∼90 amino acids in other strains). Intriguingly, while PB1-F2 is encoded by nearly all human and avian isolates, swine viruses, including S-IAV-2009, encode highly truncated forms (less than 20 residues) that are likely to lack functionality. Additional novel IAV gene products probably play important roles in viral replication and pathogenicity Citation[4,5]; it is to be expected that IAV makes the most of its extremely limited coding capacity.
In came S-IAV-2009
When multiple virions productively infect a single cell, reassortment of gene segments between viruses in progeny is common. Pandemic viruses typically arise from the recombination of animal and human viruses, generating a virus with animal glycoprotein genes on a background of mostly, or entirely, human genes.
The genetic makeup of S-IAV-2009 is complex, containing genes derived from human (PB1), avian (PA and PB2), classical swine (HA, NP and NS) and Eurasian swine (NA and M) IAVs Citation[6–8]. Despite their disparate origins, each of the genes appears to have circulated in swine for at least 10 years Citation[7]. While the virus most probably jumped directly from pigs to man, it is impossible to rule out a bridging host. Owing to the relatively limited surveillance of swine IAVs, it is possible that the reassorted virus circulated in swine for years.
Swine IAVs routinely infect humans, although prior to this year, the CDC only reported one to two annual cases. Most cases of human infection with swine IAVs are probably unreported since symptoms are indistinguishable from the spectrum of symptoms caused by human IAV infections Citation[9]. The precise moment when S-IAV-2009 crossed the species will never be known. One of the earliest cases was reported in La Gloria, Mexico in February 2009, and further genetic analysis of individual viral isolates predicts a common ancestor virus that dates back to January 2009 Citation[10].
Immunity to S-IAV-2009
The immune response to IAV is comprised of multiple elements of innate and adaptive immunity. While the innate immune system plays an essential role in limiting viral replication in the first days of infection, it is relatively nonspecific, and has yet to be rationally manipulated to provide protection against infection. Rather, vaccines are based on the induction of B- and T-cell responses. CD8+ T cells may contribute to decreasing human IAV mortality (as has been extensively documented in mice Citation[11], and even chickens Citation[12]), but as yet are unproven vaccine targets. Rather, vaccines are designed to elicit anti-IAV antibodies (Abs).
Three IAV proteins elicit robust Ab responses during infection: NP, NA and HA. Although NP is expressed on cell surfaces and can be used to target cells for destruction Citation[13,14], anti-NP Abs have never been shown to play an important role in immunity to IAV. NA-specific Abs prevent virus release from cells and limit transmission. HA-specific Abs block entry into cells by blocking virus attachment or fusion with cellular membranes, and these Abs are the only effector function of the immune system that prevents IAV infection. Consequently, HA is by far the most important IAV protein from the standpoint of epidemiology and vaccination. M2 is less immunogenic, but is a promising target for cross-protective vaccination, since its short extracellular target domain is highly conserved, and Abs can protect against lethal challenge Citation[15]. This contrasts with HA and NA, which rapidly evolve antigenically under Ab pressure. The process of antigenic evolution that results from the accumulation of amino acid substitutions is known as ‘antigenic drift’. More radical changes in HA (or NA) antigenicity, ‘antigenic shift’, occurs when an entirely new gene is introduced into the human population from the animal reservoir.
Owing to the importance of IAV as a human pathogen, and the central role of HA in IAV ecology in humans, HA has recorded many ‘firsts’. These include: the first viral protein to have its structure determined by crystallography; the first protein to be mapped antigenically with monoclonal Abs (mAbs); and the first to have its membrane fusion mechanism dissected.
Hemagglutinin is a homotrimer, with each monomer consisting of two disulfide-linked subunits, HA1 and HA2, which are generated by cleavage in the late secretory compartment during HA biogenesis . Cleavage is required to activate HA fusion activity, which is based on the low pH-induced exposure of a highly hydrophobic peptide at the HA2 NH2 terminus. Abs with potent neutralizing activity are specific for the globular domain formed by loops of the HA1 chain at the tip of the HA spikes. Most anti-HA Abs neutralize by preventing viral binding to sialic acid-bearing cellular receptors Citation[16]. A subset of Abs inhibit the fusion step of virus entry by binding either HA2 or membrane proximal regions of HA1. Fusion-inhibiting Abs are potentially important in providing protection between drifted, or even shifted, strains, since the targeted domains are more highly conserved than the globular regions Citation[17,18]. At the same time, the conservation of these domains suggests that such Abs exert little pressure in nature and, indeed, fusion-inhibiting Abs are typically far less efficient than receptor-blocking Abs at neutralizing virus.
H1: HA antigenicity in high definition
As luck would have it, thanks to Walter Gerhard and his disciples at the Wistar Institute, the H1 HA is probably the most extensively characterized antigen in existence. The ‘first’ mAbs mentioned previously were generated by Walter et al. against the A/PR/8/34 HA Citation[19], and were followed by hundreds more over the course of his career. These mAbs were used to invent ‘epitope mapping’, a three-step process Citation[19]:
• Step one: propagate virus in the presence of sufficient Ab to neutralize all wild-type virus, resulting in the selection of escape mutants, which demonstrate at least 100-fold decreased affinity for the selecting mAb Citation[20];
• Step two: sequence mutants by nascent nucleic acid sequencing technology Citation[21,22];
• Step three: use mutants to map the epitopes of a large mAb panel by direct binding assay.
Epitope mapping revealed that the H1 HA possesses four physically distinct antigenic sites designated Sa, Sb, Ca and Cb Citation[22]. The ‘S’ in Sa and Sb refers to ‘strain-specific’, as these sites are highly variable between different stains. The ‘C’ in Ca and Cb refers to ‘cross-reactive’, as these sites are positioned more proximally to the viral membrane and are more conserved between different viral strains. The ‘a’ and ‘b’ designate physically distinct sites.
Operationally, the antigenic sites are distinct, based on a large number of mAbs whose binding is diminished by mutations in a single site. Physically, the Sa and Sb sites are situated at the top of the globular domain, on the upper ridge of the sialic acid-binding pocket . The Ca site is just below the receptor-binding site and sits astride the trimer interface, subdividing the site into Ca1 and Ca2 subsites. The Cb site resides further down the spike. Each of the sites are formed by residues from different loops of the HA and are exquisitely sensitive to denaturation. Consequently, despite the initial hoopla Citation[23,24], it is apparently not possible to use short synthetic peptides corresponding to linear HA sequences to elicit Abs that bind to the globular domain with sufficient affinities to enable effective vaccination.
Drift: a well-defined mystery
The typical immune response to HA probably contains thousands of different Abs with neutralization activity Citation[25]. How then can HA escape neutralization? Although each Ab interacts with HA in a unique physiochemical manner, they share many common features of recognition, and they are similarly negatively affected by amino acid substitutions. Retrospective analysis of Gerhard et al.Citation[21] reveals that on average single amino acid substitutions in escape mutants block binding of approximately half of the Abs specific for a given site . If individuals make a balanced neutralizing response against each of the four sites, then eight (or more) mutations would be needed to generate a mutant capable of escaping all Abs. Since the frequency of escape mutants with single amino substitutions is on the order of 105Citation[26], anything beyond a double substitution (frequency 1010) is essentially impossible during the course of infection of one individual, given the amount of virus replication required (at least 1015 virions; a large number, considering each cell produces ∼103 progeny virions, and humans only have ∼1013 cells).
This leads to two distinct possibilities. First, some humans may mount dominant Ab responses against individual antigenic sites, leading to sequential selection of variants and gradual antigenic drift Citation[27,28]. Second, some amino acid substitutions may confer resistance to neutralization by a mechanism independent of their antigenic modifications. Indeed, growing IAV in the presence of a mixture of mAbs chosen to provide equal neutralization pressure on all four sites selected absorptive variants Citation[29], which are mutants with higher affinity for cellular sialic acid receptors with extremely limited antigenic alterations Citation[30].
Could such absorptive mutants be selected in vivo by individuals with intermediate levels of neutralizing Abs? This might explain why antigenic evolution in humans is punctuated rather than gradual Citation[31,32]. It might also help explain why approximately 30% of HA substitutions are located outside of defined antigenic sites Citation[32], if such mutations modulate HA-binding affinity. Clearly, a great deal remains to be learned about antigenic drift, including why IAV drifts much more rapidly in humans than other species, and why other human viruses with similar mutation rates and receptors (such as parainfluenza viruses) are essentially antigenically stable.
A silver lining to the 1976 swine vaccine debacle?
In the last century, pandemic influenza has always been associated with antigenic shifts. S-IAV-2009 is a H1N1 virus, and H1N1 viruses have been circulating since their reintroduction into the human population in 1977 (probably from a laboratory freezer, since the 1977 H1N1 virus was nearly identical to a 1950 H1N1 strain). S-IAV-2009, however, might break the ‘shift rule’, since swine viruses are quite antigenically distinct from other H1N1 viruses. S-IAV-2009 is no exception, as at least 13 amino acid substitutions in the HA and four in the NA have not been observed in previous human H1N1 viruses Citation[33]. Comparison of the S-IAV-2009 with the H1N1 component of the current vaccine (A/Brisbane/59/07) revealed only 56 and 38% sequence homology within the antigenic sites of HA and NA, respectively Citation[33]. As vaccine strains sometimes need to be modified after only one or two HA amino acid substitutions, it is unlikely that the current vaccine will afford any protection against S-IAV-2009 infection. Indeed, ferret antisera against a 2009 seasonal H1N1 human IAV did not react with S-IAV-2009 in HA-inhibition assays Citation[6]. Further support for the pandemic potential of S-IAV-2009 comes from the events of 1947, when a significant alteration in H1 antigenicity resulted in a complete vaccine failure that probably would have led to a pandemic had the novel virus not exhibited low pathogenicity Citation[34].
The transmission of S-IAV-2009 in the USA may be mitigated, however, by the 1976 swine vaccination campaign that vaccinated more than 40 million US citizens before being terminated due to the occurrence of Guillian–Barré syndrome in approximately one in 100,000 vaccinees (see Citation[103] to download a fascinating history). Plasma cells are extremely long lived, as spectacularly documented by the recovery of neutralizing Abs specific for 1918 HA from nonagenarian survivors of the epidemic Citation[35]. It will be of great interest to correlate 1976 vaccination with the presence of anti-swine HA antibodies and protection against S-IAV-2009.
Bioinformatics: no crystal ball
It is very likely that S-IAV-2009 will become established in the human population due to its antigenic novelty, and that massive amounts of vaccine will be necessary for the coming flu seasons. There is every reason to believe that S-IAV-2009 will behave like every other known human IAV and begin to drift antigenically, necessitating constant vaccine reformulation. Given the enormous progress in sequencing and bioinformatics, is it possible to predict the direction of drift and maintain a lead on generating vaccines? In a nutshell: no.
Approximately 50 amino acids contribute to HA antigenicity, and point mutations in the corresponding codons can introduce multiple potential substitutions (limited by both the genetic code and the structural flexibility of HA in maintaining function with a given substitution), leading to a daunting number of potential structures. Attempts to empirically predict drift by in vitro selection with antiserum have failed miserably Citation[30]. The reintroduction of the 1950s H1N1 virus in 1977 led to a unique natural experiment: how would drift compare during two independent trials in humans? The answer is not at all Citation[36].
There is some good news, at least in the near term. First, drift in S-IAV-2009 seems to be on the languid side compared with other human IAVs Citation[31,37]. Second, S-IAV-2009 lacks several molecular motifs associated with high transmission and pathogenesis. Unlike virtually all human isolates, S-IAV-2009 encodes Lys in place of the normal Glu at position 627 of PB2. It was recently reported that K627 is important for efficient viral transmission, by allowing the virus to replicate at lower temperatures in the upper airway Citation[38]. Third, PB1-F2 protein, a defined pathogenesis factor in mouse models Citation[39–41], is truncated in S-IAV-2009 to the point of functional irrelevance. Fourth, S-IAV-2009 is sensitive to the NA inhibitors oseltamivir and zanamivir.
The bad news is that S-IAV-2009 is probably just a handful of mutations (or a recombination event or two) from becoming highly pathogenic. Should this happen, mass vaccination will become urgent, as the absence of pre-existing immunity could result in rapid and extensive transmission. The financial price alone of this possibility will dwarf the costs of maximizing investments in monitoring, vaccine development and IAV research.
Table 1. Antigenic analysis of A/PR/8/34 HA.
Financial & competing interests disclosure
The authors are generously supported by the Division of Intramural Research, NIAID. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Swine influenza A (H1N1) infection in two children – southern California, March–April 2009. MMWR58(15), 400–402 (2009).
- Clark T, Stephenson I. Influenza A/H1N1 in 2009: a pandemic in evolution. Expert Rev. Vaccines8(7), 819–822 (2009).
- Conenello GM, Palese P. Influenza A virus PB1-F2: a small protein with a big punch. Cell Host Microbe2(4), 207–209 (2007).
- Wise HM, Foeglein A, Sun J et al. A complicated message: identification of a novel PB1-related protein translated from influenza A segment 2 mRNA. J. Virol.83(16), 8021–8031 (2009).
- Zhirnov OP, Poyarkov SV, Vorob’eva IV, Safonova OA, Malyshev NA, Klenk HD. Segment NS of influenza A virus contains an additional gene NSP in positive-sense orientation. Dokl. Biochem. Biophys.414(1), 127–133 (2007).
- Garten RJ, Davis CT, Russell CA et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science325(5937), 197–201 (2009).
- Smith GJ, Vijaykrishna D, Bahl J et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature459(7250), 1122–1125 (2009).
- Dawood FS, Jain S, Finelli L et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med.360(25), 2605–2615 (2009).
- Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans: a review of the literature. Clin. Infect. Dis.44(8), 1084–1088 (2007).
- Fraser C, Donnelly CA, Cauchemez S et al. Pandemic potential of a strain of influenza A (H1N1): early findings. Science324(5934), 1557–1561 (2009).
- Stambas J, Guillonneau C, Kedzierska K, Mintern JD, Doherty PC, La Gruta NL. Killer T cells in influenza. Pharmacol. Ther.120(2), 186–196 (2008).
- Seo SH, Webster RG. Cross-reactive, cell-mediated immunity and protection of chickens from lethal H5N1 influenza virus infection in Hong Kong poultry markets. J. Virol.75(6), 2516–2525 (2001).
- Yewdell JW, Frank E, Gerhard W. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J. Immunol.126, 1814–1819 (1981).
- Staerz UD, Yewdell JW, Bevan MJ. Hybrid antibody-mediated lysis of virus-infected cells. Eur. J. Immunol.17(4), 571–574 (1987).
- Gerhard W, Mozdzanowska K, Zharikova D. Prospects for universal influenza virus vaccine. Emerg. Infect. Dis.12(4), 569–574 (2006).
- Knossow M, Skehel JJ. Variation and infectivity neutralization in influenza. Immunology119(1), 1–7 (2006).
- Ekiert DC, Bhabha G, Elsliger MA et al. Antibody recognition of a highly conserved influenza virus epitope. Science324(5924), 246–251 (2009).
- Sui J, Hwang WC, Perez S et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol.16(3), 265–273 (2009).
- Yewdell JW, Gerhard W. Antigenic characterization of viruses by monoclonal antibodies. Annu. Rev. Microbiol.35, 185–206 (1981).
- Gerhard W, Webster RG. Antigenic drift in influenza A viruses. I. Selection and characterization of antigenic variants of A/PR/8/34 (HON1) influenza virus with monoclonal antibodies. J. Exp. Med.148, 383–392 (1978).
- Gerhard W, Yewdell J, Frankel ME, Webster R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature290(5808), 713–717 (1981).
- Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell31(2 Pt 1), 417–427 (1982).
- Green N, Alexander H, Olson A et al. Immunogenic structure of the influenza-virus hemagglutinin. Cell28(3), 477–487 (1982).
- Muller GM, Shapira M, Arnon R. Anti-influenza response achieved by immunization with a synthetic conjugate. Proc. Natl Acad. Sci. USA79(2), 569–573 (1982).
- Staudt LM, Gerhard W. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. I. Significant variation in repertoire expression between individual mice. J. Exp. Med.157(2), 687–704 (1983).
- Yewdell JW, Webster RG, Gerhard WU. Antigenic variation in three distinct determinants of an influenza type A haemagglutinin molecule. Nature279(5710), 246–248 (1979).
- Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Ann. Rev. Immunol.8, 737–771 (1990).
- Wang ML, Skehel JJ, Wiley DC. Comparative analyses of the specificities of anti-influenza hemagglutinin antibodies in human sera. J. Virol.57(1), 124–128 (1986).
- Yewdell JW, Caton AJ, Gerhard W. Selection of influenza A virus adsorptive mutants by growth in the presence of a mixture of monoclonal antihemagglutinin antibodies. J. Virol.57(2), 623–628 (1986).
- Fazekasd S. Evolution and hierarchy of influenza viruses. Arch. Environ. Health21(3), 293–303 (1970).
- Smith DJ, Lapedes AS, de Jong JC et al. Mapping the antigenic and genetic evolution of influenza virus. Science305(5682), 371–376 (2004).
- Nelson MI, Simonsen L, Viboud C et al. Stochastic processes are key determinants of short-term evolution in influenza a virus. PLoS Pathog.2(12), e125 (2006).
- Soundararajan V, Tharakaraman K, Raman R et al. Extrapolating from sequence – the 2009 H1N1 ‘swine’ influenza virus. Nat. Biotechnol.27(6), 510–513 (2009).
- Kilbourne ED, Smith C, Brett I, Pokorny BA, Johansson B, Cox N. The total influenza vaccine failure of 1947 revisited: major intrasubtypic antigenic change can explain failure of vaccine in a post-World War II epidemic. Proc. Natl Acad. Sci. USA99(16), 10748–10752 (2002).
- Yu X, Tsibane T, McGraw PA et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature455(7212), 532–536 (2008).
- Raymond FL, Caton AJ, Cox NJ, Kendal AP, Brownlee GG. The antigenicity and evolution of influenza H1 haemagglutinin, from 1950–1957 and 1977–1983: two pathways from one gene. Virology148(2), 275–287 (1986).
- Russell CA, Jones TC, Barr IG et al. The global circulation of seasonal influenza A (H3N2) viruses. Science320(5874), 340–346 (2008).
- Van Hoeven N, Pappas C, Belser JA et al. Human HA and polymerase subunit PB2 proteins confer transmission of an avian influenza virus through the air. Proc. Natl Acad. Sci. USA106(9), 3366–3371 (2009).
- Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog.3(10), 1414–1421 (2007).
- Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol.80(16), 7976–7983 (2006).
- Steel J, Lowen AC, Mubareka S, Palese P. Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLoS Pathog.5(1), e1000252 (2009).
Websites
- Global Initiative on Sharing Avian Influenza Data http://platform.gisaid.org
- National Center for Biotechnology Information www.ncbi.nlm.nih.gov/genomes/FLU/SwineFlu.html
- The Swine Flu Affair www.iom.edu/cms/aboutiOM/4081/65926.aspx