Abstract
Altered DNA methylation is ubiquitous in human cancers and specific methylation changes are often correlated with clinical features. DNA methylation biomarkers, which use those specific methylation changes, provide a range of opportunities for early detection, diagnosis, prognosis, therapeutic stratification and post-therapeutic monitoring. Here we review current approaches to developing and applying DNA methylation biomarkers in cancer therapy. We discuss the obstacles that have so far limited the routine use of DNA methylation biomarkers in clinical settings and describe ways in which these obstacles can be overcome. Finally, we summarize the current state of clinical implementation for some of the most widely studied and well-validated DNA methylation biomarkers, including SEPT9, VIM, SHOX2, PITX2 and MGMT.
The pathway is more extensively described in the ‘Expert commentary’ section.
It is increasingly accepted that cancer is an epigenetic disease as well as a genetic disease Citation[1–3]. Indeed, some cancers seem to be principally driven by epigenetic change, such as malignant rhabdoid tumors and retinoblastoma Citation[4,5]. Therefore, the assessment of epigenetic changes, such as DNA methylation, will add essential diagnostic and prognostic information with clinical relevance. However, DNA methylation biomarkers are only slowly making their way into cancer molecular diagnostics. This review focuses on approaches and challenges for DNA methylation biomarkers in cancer therapy.
Aberrant DNA methylation & cancer
In the human genome, DNA methylation occurs almost exclusively at CpG dinucleotides. Cytosine residues in a CpG dinucleotide can be methylated at the carbon-5 position, resulting in 5-methylcytosine. The methyl groups are added by DNA methyltransferases which utilize S-adenosyl-L-methionine as the methyl group donor. Most CpG dinucleotides are distributed throughout the genome, occurring at lower frequencies than expected, and are usually methylated. However, some CpG dinucleotides are clustered in CpG islands, which can span several hundreds to several thousands of base pairs Citation[6].
CpG islands are often associated with the promoter region of a gene and these promoter CpG islands are usually unmethylated. Generally, an unmethylated promoter CpG island permits transcription initiation, whereas a methylated promoter CpG island represses promoter activity Citation[7]. There is also evidence that intragenic DNA methylation plays a role in regulating gene expression Citation[8,9]. However, in this review we will focus only on methylation changes associated with promoter CpG islands, although intragenic methylation may provide valuable biomarkers in the future.
In cancer, both global hypomethylation and localized increases of methylation are observed. Global hypomethylation (decrease of overall DNA methylation levels) affects CpG dinucleotides in repetitive elements (such as long interspersed nucleotide element-1), as well as certain gene-specific promoter CpG islands Citation[10]. Local ‘hypermethylation’ (increase of DNA methylation) has often been observed at CpG islands but may affect other regions that do not fit standard CpG island algorithms as well. It has been estimated that each tumor harbors on average several hundreds of aberrantly methylated CpG islands Citation[11], many of them promoter CpG islands.
As aberrant methylation of promoter-associated CpG islands can lead to loss of gene expression, promoter methylation can be seen as an alternative gene-inactivation mechanism to chromosomal loss and loss-of-function mutations Citation[7]. However, in contrast to DNA sequence changes, such as chromosomal loss and mutations that are essentially irreversible, the reversible nature of DNA methylation offers a therapeutic opportunity to globally restore the normal epigenome using demethylating agents.
Concepts & challenges of using DNA methylation as a cancer biomarker
A number of features make DNA methylation an attractive source of potential biomarkers. DNA methylation changes are often early events in carcinogenesis, with some already present in precancerous lesions Citation[12]. As DNA methylation is a positive readout, it can be scored even in samples with low tumor purity. These characteristics make appropriately chosen DNA methylation biomarkers suitable for early detection methodologies.
Importantly, only a small fraction of aberrantly methylated genes are driver genes whose methylation is directly related to cancer initiation or progression. Thus, the vast majority of methylated genes will be ‘passengers’ that become methylated as a consequence of being part of an epigenetically silenced domain Citation[13], or derive from the methylation of stem cell bivalent chromatin domains Citation[14,15].
There are two broad groups of applications for DNA methylation analysis in cancer. First, an already diagnosed tumor can be analyzed for a range of diagnostic, prognostic and predictive methylation biomarkers. A large number of techniques are available Citation[16], although it is important to choose at least a semi-quantitative methodology. Second, DNA methylation has the potential to be used as a biomarker to confirm the presence of a tumor (or not) as in early detection and minimal residual disease detection. Such applications require sensitive methods to score the methylation status of a given sample.
Unlike mutations, which can be precisely defined, methylation takes place over a larger and often poorly defined region. In many cases, the critical informative region for the methylation analysis is unknown. Therefore, the analysis of single CpG dinucleotides is rarely suitable and is further complicated as DNA methylation patterns are often heterogeneous. Furthermore, due to sample quality limitations, the region analyzed for methylation often necessarily comprises relatively few CpG dinucleotides, which serve as a surrogate for the methylation status of the whole region. Technical limitations in PCR amplicon design, and in particular primer placement, also place constraints on the region that is analyzed.
In mutation screening, pathological aberrations are found by comparing the sequence under investigation to the corresponding wild-type sequence. However, for DNA methylation analysis, a baseline needs to be defined for every region in the appropriate normal control tissues. For some promoter CpG islands, zero methylation can be used as a baseline. However, other promoter CpG islands can show variable background methylation in healthy individuals, such as the DAPK1 promoter Citation[17], and still others show striking inter-individual variation with only some individuals showing methylation Citation[18]. Furthermore, many gene loci also show an age-dependent increase in DNA methylation Citation[19]. The latter considerations are probably most important for the use of aberrant methylation in early detection or detection of minimal residual disease, as it is expected that the methylation levels might be low and only present in a subset of cells.
The choice of region to be studied is one of the critical challenges to establishing a DNA methylation biomarker that is clinically useful. The investigated region should ideally fulfill the following criteria: first, the region should be unmethylated in normal cases and methylated in cancer cases; and second, the methylation levels of this region should clearly allow the classification of a sample as normal or cancerous. For CpG islands, these ideal situations may be true for one region of a CpG island and not another. Careful study of the entire promoter region CpG island is necessary. An example of this analysis has been carried out for the MLH1 gene Citation[20].
Methodological considerations for DNA methylation biomarkers
The principal reason for the slow implementation of DNA methylation analysis in clinical diagnostics is the general lack of consensus for an agreed methodology and the widespread use of inappropriate methodologies. In particular, the possibility that different levels of methylation might be present at certain gene loci in normal tissues necessitates the need to use quantitative methods for DNA methylation biomarkers.
There are many different locus-specific DNA methylation detection methodologies described in the literature. Some methods use genomic DNA as a starting material and are based on a digestion step with methylation-sensitive restriction endonucleases prior to PCR amplification Citation[21]. Since the enzymatic digestion might be incomplete, especially when damaged DNA samples are used, there is always the risk of encountering false-positive results. In addition, enzymatic approaches are only capable of assessing relatively few CpG dinucleotides in a region unless they are multiplexed Citation[22].
The majority of approaches for DNA methylation detection utilize bisulfite-treated DNA as the PCR template to amplify the region(s) of interest Citation[23]. During bisulfite treatment unmethylated cytosines are converted to uracils, whereas 5-methylcytosines are relatively nonreactive under the reaction conditions. During PCR amplification, the uracils are replaced with thymines, and 5-methylcytosines with cytosines. Thus, an unmethylated cytosine will become a thymine in the amplicon, whereas a methylated cytosine remains as a cytosine. Different methodologies use different strategies to investigate the degree of conversion of the CpG sites within the region of interest, to provide information about the DNA methylation status Citation[16].
The need to bisulfite-treat genomic DNA is the first hurdle for reliable DNA methylation detection. The bisulfite conversion rate will be nearly complete if the bisulfite treatment is correctly performed. However, an inferior conversion rate might occur during the bisulfite treatment procedure, resulting in false-positive results. This is particularly serious where very sensitive detection methods are chosen, such as methods based on methylation-specific PCR (MSP). The risk of amplifying templates with a poor conversion rate requires approaches or methods, such as ConLight-MSP Citation[24] or sensitive melting analysis after real time methylation-specific PCR (SMART-MSP) Citation[25], capable of identifying individual samples containing poorly converted templates. Bisulfite treatment also causes extensive DNA degradation and, in some cases, has been reported to affect up to 96% of the DNA Citation[26]. However, both problems can be minimized by using commercially available kits for bisulfite treatment, which generally outperform in-house bisulfite treatment protocols.
Another problem that needs to be carefully considered when establishing a new methylation detection assay consists of PCR amplification bias towards either unmethylated or methylated alleles Citation[27]. The amplification bias can be detected by analyzing a set of DNA standards containing known ratios of methylated to unmethylated DNA Citation[28]. Different approaches have been suggested to correct for the PCR amplification bias. An amplification bias can be manipulated by adjusting the primer annealing temperature, either when using PCR primers that do not contain CpG dinucleotides Citation[29], or when using PCR primers that include limited CpG sites Citation[28].
Another hurdle for most DNA methylation detection methodologies is the limitation when heterogeneous methylation patterns (rather than homogenous ones) are present, which are common at many gene loci, such as MGMTCitation[30]. Heterogeneous methylation is the situation where multiple (epi)alleles are present, which differ in their pattern of methylated and unmethylated CpG positions Citation[30]. Meaningful DNA methylation standards for quantification of heterogeneous methylation are not feasible as every tumor may have its own repertoire of epialleles Citation[31]. Thus, the absolute quantification of heterogeneous methylation patterns cannot be achieved without digital approaches as discussed in a later section Citation[30].
Appropriate methodologies for DNA methylation detection at single gene loci, which are suitable for use in a clinical setting, should ideally:
• Provide quantitative methylation data
• Be suitable for the investigation of degraded DNA
• Be time- and cost-effective
These considerations apply both to methodologies assessing tumor DNA directly (for diagnostic, prognostic and predictive biomarkers), as well as to methodologies assessing tumor DNA indirectly in bodily fluids (for early detection and minimal residual disease detection).
DNA methylation analysis of tumors
For the analysis of tumor biopsies, quantitative or semi-quantitative methodology is essential for robust DNA methylation analysis. Less sensitive methodologies may be used, as long as their dynamic range is able to span the potential range of tumor purities. The methods need to have a minimum analytical sensitivity of at least 5–10% as some biopsies have relatively low proportions of tumor cells. Conventional MSP, which has been widely used in methylation analysis, is unsuitable because of its propensity for artifacts and its inability to distinguish background levels of methylation from high levels of methylation. However, quantitative adaptations of MSP can be suitable (discussed in more detail in the ‘DNA methylation analysis of bodily fluids’ section). Other appropriate techniques are reviewed below.
The majority of solid tumor samples available for clinical studies are archived as formalin-fixed, paraffin-embedded (FFPE) specimens. DNA extracted from FFPE samples is invariably degraded and also prone to generate PCR artifacts caused by chemical modifications to the DNA Citation[32]. Therefore, suitable DNA methylation detection methodologies are needed for the investigation of such challenging material. However, the advantages of FFPE material are the low costs for storing FFPE blocks, the easy handling for cutting tissue sections and the ready identification of tumor-enriched areas for enrichment by macro- or micro-dissection. The latter argument is of considerable importance because most tumor tissues are contaminated with normal cells (e.g., normal tissue and infiltrating white blood cells).
Bisulfite pyrosequencing is a time- and cost-effective approach for DNA methylation detection at a single gene locus Citation[33]. This sequencing-by-synthesis approach utilizes PCR products, which have been amplified before the actual methylation analysis. The data is displayed as an average methylation level for each CpG position analyzed across all amplicons generated during PCR amplification. Bisulfite pyrosequencing delivers reliable quantitative methylation data down to approximately 5–10% for each CpG position analyzed Citation[34,35]. The software provided by the manufacturer offers the opportunity to quality control for sufficient bisulfite conversion. Pyrosequencing has a read length of up to 100 bp and is well suited for the analysis of FFPE-derived DNA Citation[33].
Methylation-sensitive high-resolution melting (MS-HRM) is another useful method for rapidly deriving methylation information as well as an attractive front-end method for bisulfite pyrosequencing Citation[36]. MS-HRM is a sensitive methylation screening approach that allows the detection of methylation down to levels of 1–5% for homogeneous methylation patterns via melting curve analysis Citation[37,38]. It also allows the visualization of heterogeneous methylation. Samples that have been identified as positive for methylation can be subjected to bisulfite pyrosequencing for quantitative DNA methylation analysis Citation[31]. Another advantage of MS-HRM is that it is a closed-tube method that allows PCR amplification and methylation analysis in one tube, without the need to remove the PCR products for further analysis. Closed-tube approaches are of particular interest to molecular diagnostics as they minimize the risk of PCR carry-over and cross contamination.
MassARRAY® (EpiTYPER®; Sequenom Inc.,) methodology allows (semi-)quantitative methylation detection determined from fragment masses after base-specific cleavage utilizing MALDI-TOF mass spectrometry Citation[39]. Methylation levels are determined as an average for a single CpG dinucleotide, or for multiple CpG dinucleotides across all amplicons generated during PCR amplification, while the analytical sensitivity is similar to that of bisulfite pyrosequencing Citation[40]. However, EpiTYPER allows the investigation of longer amplicons of up to 500 bp. This is not necessarily always an advantage in cancer studies, since clinical material for methylation analysis is often heavily fragmented.
A promising array-based approach is the Infinium HumanMethylation® 450K BeadChip microarray platform (Illumina), which can determine the methylation status of approximately 485,000 CpG dinucleotides strategically placed throughout the genome Citation[41]. The analytical sensitivity is approximately 10–20%. However, the disadvantage of a decreased sensitivity might be compensated by the fact that CpG dinucleotides from very large numbers of genes can be analyzed in parallel.
Currently, this approach requires high-quality, high-molecular-weight DNA for the whole-genome amplification step, which makes the method unsuitable for most studies where DNA comes from FFPE specimens. The development of a modified sample preparation protocol enabled the use of FFPE-derived DNA Citation[42], but the obtained methylation data still lack the reliability required for use in a clinical setting Citation[43].
Finally, amplicon-based approaches using massively parallel sequencing have been successfully applied for methylation analysis of selected loci in leukemia and lymphoma Citation[44], as well as breast cancer Citation[45]. This methodology offers the opportunity to analyze multiple DNA methylation biomarkers in parallel, making this approach relatively cost effective. However, a potential drawback of this promising approach is the risk of PCR amplification bias leading to an alteration of the allelic ratios.
DNA methylation analysis of bodily fluids
The collection of bodily fluids is often a relatively noninvasive procedure enabling early cancer detection or detection of minimal residual disease post-treatment. These fluids include plasma or serum from blood, as well as organ-specific fluids such as bronchial aspirate, sputum, ductal lavages, urine and stool.
DNA methylation biomarkers are attractive for the analysis of bodily fluids, particularly in the early detection scenario where even a large panel of mutational biomarkers will not be sensitive enough to detect the majority of cases of a given tumor type. On the other hand, relatively small panels of DNA methylation biomarkers will often be enough to cover most cases of a given tumor type Citation[46].
However, methodologies with the sensitivity and specificity suitable for the investigation of bodily fluids are particularly challenging. For detection and minimal residual disease studies, biomarkers and methods with high analytical sensitivity and, in particular, high analytical specificity need to be adopted to allow the detection of only few cancer-specific DNA methylation changes (generally methylated alleles) in what is often a large background of normal DNA (generally unmethylated alleles). Therefore, the choice of a ‘clean’ biomarker with no or very low background methylation is preferable to avoid an increased false-positive rate resulting in decreased analytical specificity.
Conventional MSP is a very sensitive methylation detection method and is frequently used to detect low levels of DNA methylation Citation[47]. MSP utilizes PCR primers that contain multiple CpG dinucleotides, therefore selecting for the specific amplification of methylated templates. However, MSP is unsuitable for use in a clinical setting. First, conventional MSP is purely a qualitative approach and delivers just a ‘yes’ or ‘no’ answer for the methylation status. Second, conventional MSP is prone to false-positive as well as false-negative results, especially when FFPE-derived DNA is analyzed Citation[48,49]. MSP is also difficult to standardize between laboratories Citation[50].
Quantitative MSP-based approaches, such as MethyLight Citation[51] or SMART-MSP Citation[25] are potentially suitable for use in molecular diagnostics as these approaches allow the selection of methylation thresholds. In addition, SMART-MSP offers the opportunity to identify incomplete bisulfite conversion which, as mentioned, can be an important source of false positives.
Digital methodologies for DNA methylation detection
An increasingly important set of quantitative methodologies are based on digital PCR. For these approaches, samples are diluted to the point where the majority of wells contain either no template or only a single template copy, and the subsequent PCR amplification of single templates results in a homogeneous amplicon population. Individual wells that are positive for PCR amplification can be analyzed by Sanger sequencing Citation[52], although high-resolution melting analysis is very useful prior to Sanger sequencing since it allows the rejection of wells containing primer-dimers and wells where the amplicon population originated from more than one template copy. Furthermore, as unmethylated sequences can be identified, only methylated sequences need to be sequenced, therefore greatly reducing sequencing costs Citation[53].
Importantly, limiting dilution approaches are free of PCR amplification bias, and therefore accurately reflect the amount of methylated alleles in a sample. However, the drawback of these methodologies is the need to analyze hundreds of wells to get the analytical sensitivity required. The requirement to analyze such an amount of wells can make digital approaches based on standard PCR apparatus relatively expensive.
Methyl-BEAMing (beads, emulsion, amplification and magnetics) is another digital approach in which PCR amplification occurs on single beads in an emulsion of microdroplets, followed by probes for DNA methylation detection Citation[54]. This approach is currently best suited for homogeneously methylated loci as only probes for unmethylated or fully methylated templates are currently used.
The need to achieve high analytical sensitivity, even for heterogeneously methylated loci, is met by massively parallel sequencing Citation[44,45]. However, the use of pregenerated PCR products might skew the analysis owing to PCR amplification bias. Notably, PCR amplification bias will no longer be an issue for third-generation sequencing, where single unamplified molecules can be sequenced and methylation read directly Citation[55,56].
Bioinformatic concepts & tools
The development of DNA methylation biomarkers involves several bioinformatic and statistical steps, which have a strong influence on a biomarker’s accuracy and robustness. While it is strongly advisable to have an experienced biostatistician involved in the process of biomarker discovery and validation, a number of software tools have been developed that facilitate key steps of biomarker development. Here we focus on tools for the late stages of biomarker development, namely on assay design, data normalization, statistical analysis, threshold selection and statistical validation. More comprehensive descriptions of the bioinformatic concepts and tools for biomarker development, and the analysis of DNA methylation data in a broader context have been published Citation[57,58].
Software tools supporting assay design are available for several widely used methods of locus-specific DNA methylation analysis. For example, MethPrimer, Citation[59], Methyl Primer Express® (Life Technologies) Citation[201] and BiSearch Citation[60], facilitate the design and validation of primers that are tailored to bisulfite-converted DNA, and MethMarker Citation[61] supports assay design for an array of different methods, including bisulfite pyrosequencing, combined bisulfite restriction analysis and locus-specific methylated DNA immunoprecipitation.
A second class of software tools supports locus-specific data normalization and analysis. For clonal bisulfite sequencing, BiQ Analyzer Citation[62] and QUMA Citation[63] provide an interactive and user-friendly workflow that includes alignment to the genomic reference sequence, identification of methylation-specific C to T ‘polymorphisms’ and estimation of site-specific DNA methylation levels. The BiQ Analyzer HT software extends this concept to deep sequencing of bisulfite-converted DNA Citation[64], for example, on the Roche 454™ or the Ion Torrent PGM™ (Life Technologies) sequencers. Similar analysis pipelines also exist for EpiTYPER Citation[65] and combined bisulfite restriction analysis Citation[66].
Finally, for any newly developed biomarkers it is necessary to select appropriate thresholds for the DNA methylation levels and to quantitatively evaluate their performance in terms of sensitivity, specificity and robustness. Currently, only the MethMarker software provides direct support for this task Citation[61], while most researchers use general-purpose tools such as the R statistics package for performing this type of analysis in highly customized ways Citation[202]. It is anticipated that more comprehensive and standardized toolboxes for epigenetic biomarker development will emerge over time, as methods and concepts become increasingly standardized.
DNA methylation biomarkers for clinical use
In this final section, we review biomarkers at various stages of development and/or clinical implementation for a range of clinical scenarios. We start off with a consideration of the emerging field of constitutional methylation, which requires highly sensitive and specific quantitative assays, and then discuss biomarkers for the early detection of colorectal, lung and prostate cancer. A discussion of prognostic biomarkers is followed by a discussion of predictive biomarkers followed by a brief section on monitoring the effects of epigenetic therapies.
Risk stratification: constitutional methylation & cancer predisposition
Constitutional methylation describes the situation where, in some individuals, DNA methylation is seen throughout the normal somatic cells of one or more tissues in an allelic fashion, often in a mosaic form Citation[67]. Where it affects a driver gene, such as one of those underlying familial cancer, it is a conceivable source of cancer predisposition. This was first reported for the MLH1 gene in colorectal cancer where an individual with early-onset colorectal cancer had one of the alleles methylated in the peripheral blood and loss of the remaining normal allele in the tumor Citation[68].
Subsequently, in studies of the BRCA1 and MGMT genes, it was shown that constitutional methylation could be present in a mosaic form in the peripheral blood, and often at allelic frequencies of less than 1% Citation[18,69]. A detailed study by Wong et al. showed that such constitutional BRCA1 methylation was found in the peripheral blood leukocytes of more than 30% of the women with BRCA1-like breast cancers, whereas it was only present in a low percentage (3–4%) of the controls and the women with cancers that were not BRCA1-like Citation[70]. The BRCA1-like breast cancers were methylated for BRCA1, consistent with direct causation.
However, the study of constitutional methylation is still in its early stages and is confined to research laboratories. As individuals at high risk of developing cancer may have an opportunity to lower their risk of cancer development through dietary or pharmaceutical interventions, it is conceivable that screening may be implemented in the future.
As blood can be collected relatively noninvasively it is the ideal tissue for assessing constitutional methylation, particularly of multiple genes. However, methylation for any one gene may be less than in the target tissue for the corresponding cancer. Thus the investigation of low-level constitutional methylation faces similar methodological challenges as have been described for the investigation of DNA methylation for early detection, the main difference being that constitutional methylation is assessed in the cellular fraction of blood whereas early detection is generally in a noncellular fraction. Therefore, both situations share the same considerations for choosing the appropriate approaches for DNA methylation detection.
Constitutional methylation can create a problem for some early detection strategies. A biomarker like MGMT, which is methylated at low levels in some individuals, could mistakenly be considered as a positive for early detection and should thus be used with caution.
Biomarkers for early detection of cancer
Achieving early detection of the onset of cancer is a major goal of cancer research. Early detection offers the opportunity for therapeutic intervention at an early stage of cancer development, which is considered to be associated with a better disease outcome. The early presence of aberrant DNA methylation, even in (pre)cancerous lesions, makes the use of DNA methylation biomarkers an attractive possibility for early detection. Indeed, the majority of commercially available DNA methylation biomarker-based assays are thus directed towards early detection .
Colorectal cancer
Early detection of colorectal cancer has been achieved by highly invasive methods like colonoscopy, or by less-invasive methods of poor sensitivity and specificity like the fecal occult blood test. Analysis of plasma or stool for genes that become methylated early in colorectal cancer development has recently been developed as an alternative.
Epigenetic silencing of the septin 9 (SEPT9) gene by promoter methylation in plasma has been shown to be a promising biomarker for detection of colorectal cancer with improved sensitivity and specificity compared to the current fecal occult blood and DNA testing. For example, a blinded study of plasma SEPT9 methylation showed a sensitivity of 67% and specificity of 89% for the detection of colorectal cancer in patients Citation[71]. Aberrant SEPT9 methylation was also detectable in approximately 30% of patients with precancerous lesions (adenomas and hyperplastic polyps) Citation[72], indicating the potential of SEPT9 methylation testing for early detection of colorectal cancer. However, a recent study concluded that SEPT9 alone might not be sufficient to detect precancerous lesions Citation[73].
Another DNA methylation biomarker for the early detection of colorectal cancer is the vimentin (VIM) gene. VIM was chosen as a biomarker because it was methylated in colorectal cancers but not in normal colorectal mucosa or other healthy tissues. It was shown that it was readily detectable in stool and became the basis of the first commercially available DNA methylation-based diagnostic test Citation[74,75]. At a diagnostic specificity cut-off of 95%, methylation analysis for VIM from sample tissues allowed the detection of 86% of colon cancers and 76% of adenomas Citation[76]; a lower sensitivity would be expected in plasma.
Lung cancer
Over 75% of lung cancer patients are diagnosed at a locally advanced stage or with metastatic disease, partially due to the lack of an effective screening program for early detection. The presence of a smoking-related, high-risk group makes screening a highly attractive strategy for early detection of lung cancer. Thus, molecular biomarkers, especially occurring at early stages of lung tumorigenesis, have been examined for early detection of lung cancer. Currently, the most promising methylation biomarkers are CDKN2A and SHOX2.
CDKN2A (p16) was the first tumor suppressor gene found to be inactivated by promoter methylation in lung cancer Citation[77]. Transcriptional silencing of CDKN2A by promoter methylation is an early event during lung tumorigenesis, regardless of histological subtype. In addition, CDKN2A methylation has been detected in lung cancer patients from both smokers and nonsmokers Citation[78], although smoking seems to be positively associated with CDKN2A methylation Citation[79]. Two early studies examined the potential of CDKN2A methylation in sputum samples as a biomarker for early detection of individuals at high risk of developing lung cancer Citation[80,81]. Both studies found that CDKN2A methylation was detectable in sputum from patients diagnosed with squamous cell carcinoma of the lung, not only at the time of diagnosis but also before the clinical diagnosis. Subsequent studies have shown that aberrant CDKN2A methylation is also detectable in other body fluids of lung cancer patients, including plasma, serum and bronchoalveolar lavage Citation[82–84].
Methylation of SHOX2 is an emerging biomarker for diagnosis of lung cancer, particularly in cases where histology and cytology results give an inconclusive diagnosis. A quantitative assessment of SHOX2 methylation status in bronchial aspirates helped to distinguish between malignant lung cancer and several benign lung diseases Citation[85]. In particular, SHOX2 methylation was found in 62% of bronchial aspirates that were negative by cytology. Furthermore, a significant correlation between SHOX2 methylation and copy number gain by amplification was seen in lung tumors, facilitating the detection of SHOX2 methylation in clinical samples Citation[86]. For SHOX2 methylation in plasma samples, a sensitivity of 60% and a specificity of 90% has been reported for detecting lung cancer Citation[87].
Prostate cancer
Serum prostate-specific antigen (PSA) testing has been implemented as a screening method for detection of patients with prostate cancer for over two decades. Unfortunately, an increased PSA level is not only caused by prostate cancer but also by other noncancerous conditions such as inflammation, benign prostatic hyperplasia and prostatitis. Serum PSA testing is thus prone to generating false-positive results. It remains unclear whether PSA screening in the population has any effect on public health. Therefore, tumor-specific biomarkers that can complement standard PSA testing are in great demand for accurate early diagnosis of patients with prostate cancer.
Epigenetic silencing of GSTP1 by promoter methylation is recognized as a common event in prostate cancer. It is the most frequent somatic alteration observed in this disease (≥90% of all cases), but is rarely seen in normal prostate or benign prostatic hyperplasia tissues Citation[88]. Inactivation of GSTP1 by promoter methylation occurs early during prostate cancer tumorigenesis and has thus been detected in precancerous prostatic lesions Citation[88]. Furthermore, aberrant GSTP1 methylation is detectable in various body fluids (serum, plasma and urine) Citation[89–91]. Thus, detection of early-stage prostate cancer may be possible by examination of the GSTP1 methylation status in biopsied prostate tissues or in body fluids.
Prognostic biomarkers for cancer progression & outcome
Prognostic biomarkers are considered to provide information on the patient’s overall survival if the cancer is left untreated, as is sometimes the case after surgical resection. The identification of suitable prognostic biomarkers is particularly challenged by the requirement for large and well characterized sample cohorts. Although many single biomarkers and biomarker panels have been proposed in the literature, only a fraction have undergone any form of validation past the discovery phase.
An important exception is PITX2 methylation, which is a promising prognostic biomarker in breast and prostate cancer. Through several studies, node-negative, estrogen-receptor positive breast cancer patients with a methylated PITX2 promoter have been associated with a poor outcome without any adjuvant therapy Citation[92], as well as an increased risk of recurrence after surgery when treated with tamoxifen Citation[93,94]. A poor outcome has also been seen in node-positive, estrogen receptor-positive, HER-2/neu-negative breast cancers with a methylated PITX2 promoter treated with an anthracycline Citation[95]. Methylation of PITX2 has also been shown to be associated with a higher risk of relapse of prostate cancer patients after radical prostatectomy Citation[96,97].
Other potential prognostic methylation biomarkers are a long way from routine implementation but nevertheless are showing promise. The promoter methylation of four selected genes (CDKN2A, CDH13, RASSF1A and APC) was associated with early recurrence in stage I non-small-cell lung carcinoma Citation[98]. Interestingly, a subsequent study used this panel of methylation biomarkers to determine response to a combined therapy involving the use of a demethylating agent and a histone deacetylase inhibitor Citation[99]. The promoter methylation status in the plasma DNA was analyzed before therapy and after one cycle of therapy. Methylation for two or more of the panel of four biomarkers was predictive of a response to the epigenetic therapy. The patients with two or more methylated genes (methylation-signature-positive patients) before therapy showed a decrease in the methylation of these genes after therapy. Most of these patients had either stable disease or objective responses. However, no objective responses were observed for the methylation-signature-negative patients. This is an intriguing example of a poor prognostic biomarker becoming a good predictive biomarker.
Predictive biomarkers for cancer therapy treatment decisions
While classical pharmacogenomic biomarkers are useful in determining the toxicity of a given drug to an individual, tumor pharmacogenomic biomarkers that are not shared with the germline tissue are important in determining the therapeutic index of a given treatment. The benefit of pharmacogenomics is often limited to a relatively small group of cancer patients due to the low frequency of mutations for which targeted drugs are currently available. Alterations in the cancer epigenome are likely to be at least as important in determining the pharmacogenomic phenotype of the tumor as mutational changes. The new discipline of pharmacoepigenomics investigates variations in the cancer epigenome to determine the druggable phenotype of the tumor Citation[100]. Therefore, determining the pharmacoepigenomic as well as the pharmacogenomic phenotype of a tumor will reveal more targets for personalized cancer therapy and could help to minimize the treatment burden many cancer patients face with untargeted therapies. Moreover, treatment that is likely to be ineffective but which still puts patients at risk of potentially serious side effects can be avoided.
So far, research in pharmacoepigenomics has focused on DNA methylation changes to determine variations in chemotherapeutic drug response. Despite a promising list of biomarker candidates, few of the reports have been followed-up by subsequent studies. In many cases, conventional (nonquantitative) MSP was used to identify methylated biomarkers and in some cases this has led to erroneous results Citation[48,101].
So far, the majority of candidate predictive biomarkers belong to the group of DNA repair genes. These are often altered in cancer and can significantly influence the response to chemotherapy. Only one of these biomarkers, MGMT, has reached any level of clinical implementation. The MGMT gene encodes the DNA repair protein O6-methylguanine DNA methyltransferase, which removes alkyl groups from the O6-position of guanine residues. If MGMT is inactivated and the damage is left unrepaired, those affected cells which do not undergo apoptosis develop a mutator phenotype that drives tumor evolution. However, the repair deficiency also constitutes the major molecular weakness of the tumor, which can be exploited as a therapeutic target. Methylation of the MGMT gene renders tumors susceptible to the cell damaging effects of alkylating drugs Citation[102,103] and it has been shown that glioblastoma patients with a methylated MGMT gene show a survival benefit when treated with the alkylating drug temozolomide Citation[50].
Despite the fact that MGMT methylation is common in other tumor types, in particular colorectal cancer, temozolomide is currently used for only some tumor types, such as glioblastoma, astrocytoma and malignant melanoma Citation[104]. However, MGMT methylation testing is not routinely offered, even for glioblastoma patients. The need for routine MGMT methylation testing is partly limited by the fact that adjuvant temozolomide treatment is quite well tolerated by the patients and therefore currently given to all patients independent of the MGMT methylation status as the default option.
Multiple approaches have been developed for MGMT methylation detection (for further information, see Citation[36,105–107]). Different laboratories typically use different approaches, and while there has been only slow progress towards standardization, there is a trend towards using quantitative DNA methylation detection approaches. The use of quantitative approaches is a prerequisite as constitutional methylation at the MGMT locus in normal individuals can result in false-positive results Citation[18].
Aberrant gain of DNA methylation of other DNA repair genes has also been identified to be associated with a susceptibility or response to chemotherapeutic drugs Citation[108]. For example, patients with methylation of the WRN gene in their primary colorectal tumors showed increased sensitivity to the topoisomerase-I inhibitor irinotecan Citation[109]. Increased sensitivity of colorectal cancer cell lines to irinotecan has also been associated with methylation of UGT1A1, whose protein product is involved in drug detoxification Citation[110].
Loss of DNA repair function of genes in the BRCA–Fanconi anemia pathway via DNA methylation of BRCA1 and FANCF, has also been suggested as a chemotherapeutic target. PARP inhibitors have been proposed for the treatment of breast and ovarian cancer patients carrying BRCA1 or BRCA2 mutations Citation[111]. More recently, BRCA1 methylation was shown to sensitize breast cancer cell lines to treatment with PARP inhibitors Citation[112]. Although an increased sensitivity to cisplatin was reported in ovarian cancer cell lines if FANCF is aberrantly methylated Citation[113], FANCF methylation is rarely, if ever, seen in clinical ovarian cancer samples Citation[114]. Aberrant DNA methylation of CHFR, which codes for a protein involved in the maintenance of genome stability, has been associated with an increased sensitivity to the treatment with paclitaxel or docetaxel in gastric cancer cell lines Citation[115,116], cervical cancer cell lines Citation[117], endometrial cancer cell lines Citation[118] and in oral squamous cell carcinoma patients Citation[119]. However, the role of the methylation of CHFR in the increased sensitivity of gastric tumors to microtubule inhibitors is unclear due to two underpowered and contradictory studies Citation[116,120].
An intact apoptotic apparatus is necessary for the cytotoxic action of many DNA-damaging drugs. It has been reported that methylation of the mismatch repair gene MLH1, which also functions to promote apoptosis following DNA damage, is associated with cisplatin resistance in ovarian cancer Citation[121,122] and resistance to treatment with the antimetabolite 5-fluorouracil in colorectal cancer cell lines Citation[123]. Intriguing recent evidence also has been provided that loss of function of the pro-apoptotic DAPK1 gene by promoter methylation could be responsible for acquired resistance to the EGF receptor-targeted therapeutic drugs cetuximab and erlotinib in non-small-cell lung cancer and head and neck squamous cell carcinoma cell lines Citation[124]. This awaits confirmation in the appropriate patient cohorts.
Other reported methylated biomarkers include transcription factors whose role in chemosensitivity or chemoresistance remains poorly understood. Methylation of TP73 was associated with an increased sensitivity to alkylating drugs in a large panel of cancer cell lines Citation[125] but has not been further studied. Recently, methylation of TFAP2E was shown to be associated with chemoresistance in colorectal cancer, in particular to 5-fluorouracil-based chemotherapies. Chemoresistance is probably mediated by DKK4, an antagonist of WNT signaling, which is already known to be involved in chemoresistance to 5-fluorouracil in colorectal cancer cell lines Citation[126].
Assessing the effect of therapies with demethylating drugs
5-azacytidine and 5-aza-2´-deoxycytidine are demethylating drugs that have been approved by the US FDA for treatment of myelodysplastic syndrome. Both drugs also showed promising results at low doses in clinical trials, either as single agents or in combination with other drugs, for the treatment of other blood cancers and solid tumors Citation[127]. 5-azacytidine and 5-aza-2´-deoxycytidine are cytosine analogs, which can substitute for cytosine bases in newly synthesized DNA strands. Once incorporated into DNA, the cytosine analogs can inhibit DNA methyltransferases during the attempt to methylate cytosines in CpG dinucleotides. The inhibition of DNA methyltransferases results in reduced methylation levels during cell division Citation[127].
It is necessary to monitor DNA methylation levels at different time points to assess the genome-wide demethylating effects to patients undertaking treatments with demethylating drugs. Demethylating activity could be studied either by measuring changes to global DNA methylation levels or methylation changes at a number of individual gene loci. We need appropriate methodologies to provide quantitative data in order to provide reliable tools for monitoring the effects of demethylating drugs during a patient’s treatment course Citation[128].
Measurement of global 5-methylcytosine is often not feasible as the different approaches usually require extensive amounts of DNA, which are often not available Citation[129,130]. An alternative to measuring global DNA methylation levels is assessing changes in methylation of long interspersed nucleotide element-1 repeats Citation[129], which are widely distributed throughout the genome. This has its own problems as the use of consensus sequence PCR amplification makes it difficult to determine exactly what is being measured.
A feasible alternative is to use a panel of methylated biomarkers. Thus, if the parent tumor is methylated for one or more of the panel, those panel members can be used for subsequent monitoring of the tumor – an approach that has now been used successfully by several groups. In a cohort of patients with myelodysplastic syndrome treated with 5-aza-2´-deoxycytidine, methylation was quantitatively determined by bisulfite pyrosequencing for a panel of genes Citation[128]. Overall DNA methylation was established and validated as an independent prognostic predictor for survival. Clinical response was associated with a reduction in DNA methylation.
Expert commentary
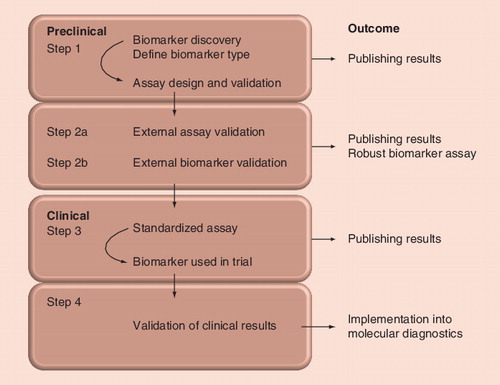
Despite general optimism about DNA methylation as a source of cancer biomarkers, and the discovery of many promising candidate genes, we are far from the clinical implementation of all but a few of those candidate genes in molecular diagnostics. There are many challenges faced by bringing any candidate biomarker from the laboratory into a clinical setting, and the majority of candidate biomarkers have not progressed past the early steps. We have outlined a pathway in , which we will use as the basis of this commentary.
Many of the biomarkers have been candidate genes that have been found to be methylated in one or more cancer types. More systematic genome-wide approaches have largely concentrated on finding the repertoire of methylated genes in a given cancer type, for example: by restriction landmark genomic scanning; differential methylation hybridization; identification of genes that are re-expressed by treatments with demethylating agents; methylated DNA immunoprecipitation; and the use of array platforms such as the Infinium HumanMethylation 27K or 450K BeadChip microarray (see step 1 of ) Citation[41,131,132].
DNA methylation biomarker discovery has rarely been followed-up with systematic development and validation strategies. Although recurrently methylated genes can all be considered as candidate biomarkers, much work needs to be done before it can be determined whether they are truly useful. To date, relatively few of the known methylated genes have been tested as potential prognostic or predictive biomarkers, and a significant portion of the work has been done in cell lines. Moreover, the work that has been performed in a clinical setting has usually been done on small numbers of patients. For genes that will be used in early detection, studies that show that methylation occurs as an early event and is restricted to cancer cells needs to be done. In all cases, robust, validated assays need to be designed. Dissemination of these results needs to occur to allow other groups the opportunity of testing the biomarkers (see step 1 of ).
External validation of the assays is very important (see step 2a of ). The uptake of DNA methylation biomarkers has often been hindered by conflicting results from different investigators. It is quite often obvious that the discrepant reports have investigated different regions of the CpG island associated with the candidate biomarker and/or used different methodologies for DNA methylation detection.
The choice of the region for DNA methylation analysis is of particular importance, since the assumption that every region of a particular CpG island contains the same biological relevance is incorrect for most candidate genes Citation[133,134]. Furthermore, constraints in PCR primer design or constraints associated with a particular methodology often dictate the choice of the region investigated rather than an evidence-driven rationale. We therefore suggest that it is essential that the methylation levels of the region chosen for DNA methylation detection are clinically informative for any particular biomarker.
Different methodologies interrogate and display the methylation levels differently and the results can only be readily compared if the same approach has been used Citation[135]. Furthermore, each approach has its strengths and weaknesses for the investigation of heterogeneous DNA methylation patterns, which adds even more complexity to the methylation analysis Citation[30]. Therefore, appropriate DNA methylation detection methodologies for single gene and multiplexed gene analysis need to be agreed on. As a minimum prerequisite, they need to deliver quantitative methylation data. This is of particular importance to define thresholds and to identify background methylation. Moreover, many of the methylation results in the literature are incorrect as they come from nonexpert laboratories using DNA methylation detection methodologies that are prone to false positives or false negatives. This is particularly the case with nonquantitative MSP-based methodologies and poorly controlled restriction endonuclease-based approaches.
Many preclinical studies are relatively small studies, and proper validation in larger sample groups is crucial (see step 2b in ). Even a large study will require external validation of the candidate methylated region as a biomarker. In the first instance, this will involve independent validation using a different cohort by the same laboratory or preferably by different laboratories, often using different methodologies Citation[136]. In addition, appropriate patient cohorts need to be characterized, and dissemination of these results in publications and in conference presentations need to occur to strengthen the case for further studies.
Once a promising DNA methylation biomarker is independently validated, and a standardized robust assay is available, it can be used as a biomarker in an appropriate retrospective or prospective trial (see step 3 of ). If the biomarker is then shown to be useful, and particularly if the results are published in an influential journal, it may then reach wider application. Further supportive data is likely to lead to demand for the assay and its implementation in a number of molecular diagnostic laboratories (see step 4 of ). For detailed flowcharts for biomarker studies, we refer the reader to the homepage of Cancer Research UK Citation[203].
The lack of standardization is currently the largest hurdle to overcome to speed up the process of implementing recognized DNA methylation biomarkers such as MGMT methylation into molecular diagnostics. Accurate correlation with clinical outcomes is difficult without reliable standardized methodologies. To some extent, this need is being met by the commercial sector which is supplying validated test kits that are becoming the de facto gold standards, often with proprietary biomarkers . However, these test kits are often expensive and thus it is desirable that other gold standards are developed via multicenter collaborations.
Finally, the use of DNA methylation biomarkers in molecular diagnostics is also impeded by the reality that very few diagnostic laboratories currently have the expertise to deal with the complexities of DNA methylation analysis. A considerable degree of sophistication is required in the interpretation of the data, and even when commercial kits are being used the interpretation of the results can be more challenging and demanding than for mutation detection. Here, an increased focus on education and dissemination of good practices for DNA methylation analysis will be necessary.
Five-year view
Both genetic and epigenetic alterations of tumors will be considered for decision-making processes in clinical practice. At the moment, molecular information is fragmentary and usually limited to a small set of genetic and epigenetic candidate loci. Much information remains to be derived from genome-wide studies of DNA methylation changes in cancer and this will become increasingly applied, leading to a greater appreciation of the role of epigenetic change in tumor development and the adoption of therapies targeting specific epigenetic changes. Thus, DNA methylation biomarkers will play an increasing role in clinical trials and ultimately in molecular diagnostics. A new breed of molecular pathologist will be required to routinely interpret complex methylation data and integrate it with mutational and gene expression data to implement predictive medicine.
Methylation profiling of tumors will be more common to assess an increasingly large number of prognostic and predictive biomarkers. Appropriate diagnostically friendly platforms will be developed to allow the simultaneous DNA methylation detection across multiple gene loci. There will be increasing use of tumor-specific DNA methylation biomarkers in bodily fluid analysis, since applications for bodily fluid analysis already have the most products on the market .
Quantitative approaches for the detection of aberrant DNA methylation will have superseded most of the nonquantitative methodologies, at least in clinical settings. Methodologies used will be compatible with degraded DNA. An array-based system suitable for DNA methylation analysis of a chosen set of biomarkers from FFPE would be desirable in terms of a clinical workflow. For early detection and the detection of minimal residual disease, digital approaches for counting methylated alleles will be used much more frequently. Next-generation sequencing, which will become central in molecular diagnostic laboratories, is the ideal platform for characterizing the complex sequences of methylated regions and may increasingly be used to count methylated alleles.
The ultimate digital approach for DNA methylation analysis will be direct single molecule sequencing of 5-methylcytosines without bisulfite treatment prior to analysis. This circumvents the danger of incomplete bisulfite conversion which can result in false-positive results. Furthermore, this approach does not require a PCR amplification step therefore avoiding amplification errors and an amplification bias towards either unmethylated or methylated sequences. Such approaches will become possible with nanopore DNA sequencing Citation[55] or single-molecule, real-time sequencing Citation[56], and other third-generation sequencing technologies.
Table 1. Commercially available tests for DNA methylation detection in clinical specimens.
Key issues
• Epigenetic changes are of equal or greater importance than mutational changes in cancer development.
• Most, if not all, cancers show aberrant DNA methylation.
• Aberrant DNA methylation of specific genomic regions occurs early in carcinogenesis.
• Two main types of DNA methylation biomarkers for cancer exist: those used to analyze tumor biopsies in order to guide therapeutic decisions; and those used to interrogate bodily fluids about the presence of cancer.
• Relatively few DNA methylation biomarkers have been developed for routine usage relative to the potential number that exists.
• Lack of methodology standardization impedes biomarker uptake.
• Quantitative methodologies are essential for the identification of clinically relevant thresholds.
• It is necessary to develop standardized methodologies based on multilaboratory quality control programs.
• It is necessary for trials to be established to validate candidate biomarkers.
• The interpretation of DNA methylation results requires expert analysis and thus testing should be performed in expert centers.
Acknowledgements
The authors thank Giada Zapparoli, Annette Lim, Dan Mellor, Darren Korbie, Jeff Craig, Nicholas Wong and Denise O’Keefe for critical reading and helpful comments on this manuscript. We acknowledge the National Breast Cancer Foundation (Australia), Cancer Australia (Lung and Breast Cancer Research Programs), the Cancer Council of Victoria, the Victorian Cancer Agency, the Susan G Komen for the Cure Foundation and the US Department of Defense Breast Cancer Research Program for funding epigenetic biomarkers projects which led directly to the consideration of the issues discussed in this manuscript.
Disclaimer
Views and opinions of, and endorsements by the authors do not reflect those of the US Army or the US Department of Defense.
Financial & competing interests disclosure
A Dobrovic is the co-inventor of intellectual property on methylation-sensitive high-resolution melting and sensitive melting analysis after real time methylation-specific PCR held by the Peter MacCallum Cancer Centre (Australia) and the University of Aarhus (Denmark). T Mikeska is the co-inventor of intellectual property on approaches for the detection of MGMT promoter methylation in clinical samples. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No funded writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Schuebel KE, Chen W, Cope L et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet.3, 1709–1723 (2007).
- Ushijima T, Asada K. Aberrant DNA methylation in contrast with mutations. Cancer Sci.101, 300–305 (2010).
- Taby R, Issa JP. Cancer epigenetics. CA Cancer J. Clin.60, 376–392 (2010).
- McKenna ES, Sansam CG, Cho YJ et al. Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol. Cell. Biol.28, 6223–6233 (2008).
- Zhang J, Benavente CA, McEvoy J et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature481, 329–334 (2012).
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature321, 209–213 (1986).
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med.349, 2042–2054 (2003).
- Jones PA. The DNA methylation paradox. Trends Genet.15, 34–37 (1999).
- Shenker N, Flanagan JM. Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br. J. Cancer106, 248–253 (2012).
- Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics1, 239–259 (2009).
- Rauch TA, Zhong X, Wu X et al. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc. Natl Acad. Sci. USA105, 252–257 (2008).
- Kanai Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci.101, 36–45 (2010).
- Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet.70, 277–308 (2010).
- Ohm JE, McGarvey KM, Yu X et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet.39, 237–242 (2007).
- Widschwendter M, Fiegl H, Egle D et al. Epigenetic stem cell signature in cancer. Nat. Genet.39, 157–158 (2007).
- Kristensen LS, Hansen LL. PCR-based methods for detecting single-locus DNA methylation biomarkers in cancer diagnostics, prognostics, and response to treatment. Clin. Chem.55, 1471–1483 (2009).
- Reddy AN, Jiang WW, Kim M et al. Death-associated protein kinase promoter hypermethylation in normal human lymphocytes. Cancer Res.63, 7694–7698 (2003).
- Candiloro IL, Dobrovic A. Detection of MGMT promoter methylation in normal individuals is strongly associated with the T allele of the rs16906252 MGMT promoter single nucleotide polymorphism. Cancer Prev. Res.2, 862–867 (2009).
- Issa JP. Epigenetic variation and human disease. J. Nutr.132, 2388S–2392S (2002).
- Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res.59, 2029–2033 (1999).
- Levenson VV. DNA methylation as a universal biomarker. Expert Rev. Mol. Diagn.10(4), 481–488 (2010).
- Sepulveda AR, Jones D, Ogino S et al. CpG methylation analysis – current status of clinical assays and potential applications in molecular diagnostics: a report of the association for molecular pathology. J. Mol. Diagn.11, 266–278 (2009).
- Frommer M, McDonald LE, Millar DS et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA89, 1827–1831 (1992).
- Rand K, Qu W, Ho T, Clark SJ, Molloy P. Conversion-specific detection of DNA methylation using real-time polymerase chain reaction (ConLight-MSP) to avoid false positives. Methods27, 114–120 (2002).
- Kristensen LS, Mikeska T, Krypuy M, Dobrovic A. Sensitive melting analysis after real time-methylation specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection. Nucl. Acids Res.36, E42 (2008).
- Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucl. Acids Res.29, E65 (2001).
- Warnecke PM, Stirzaker C, Melki JR et al. Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucl. Acids Res.25, 4422–4426 (1997).
- Wojdacz TK, Hansen LL. Reversal of PCR bias for improved sensitivity of the DNA methylation melting curve assay. Biotechniques41, 274–278 (2006).
- Shen L, Guo Y, Chen X, Ahmed S, Issa JP. Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. Biotechniques42, 48–58 (2007).
- Mikeska T, Candiloro IL, Dobrovic A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics2, 561–573 (2010).
- Candiloro ILM, Mikeska T, Dobrovic A. Assessing combined methylation-sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation. Epigenetics6, 500–507 (2011).
- Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am. J. Pathol.161, 1961–1971 (2002).
- Mikeska T, Felsberg J, Hewitt CA, Dobrovic A. Analysing DNA methylation using bisulphite pyrosequencing. Methods Mol. Biol.791, 33–53 (2011).
- Dejeux E, Audard V, Cavard C et al. Rapid identification of promoter hypermethylation in hepatocellular carcinoma by pyrosequencing of etiologically homogeneous sample pools. J. Mol. Diagn.9, 510–520 (2007).
- Lillycrop KA, Phillips ES, Torrens C et al. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR α promoter of the offspring. Br. J. Nutr.100, 278–282 (2008).
- Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucl. Acids Res.35, E41 (2007).
- Mikeska T, Dobrovic A. Methylation-sensitive high resolution melting for the rapid analysis of DNA methylation. In: Epigenetics: A Reference Manual. Craig JM, Wong NC (Eds). Caister Academic Press, Norwich, UK, 325–335 (2011).
- Wojdacz TK. Methylation-sensitive high-resolution melting in the context of legislative requirements for validation of analytical procedures for diagnostic applications. Expert Rev. Mol. Diagn.12(1), 39–47 (2012).
- Ehrich M, Nelson MR, Stanssens P et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc. Natl Acad. Sci. USA102, 15785–15790 (2005).
- Coolen MW, Statham AL, Gardiner-Garden M, Clark SJ. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: critical evaluation and improvements. Nucl. Acids Res.35, E119 (2007).
- Dedeurwaerder S, Defrance M, Calonne E et al. Evaluation of the Infinium Methylation 450K technology. Epigenomics3, 771–784 (2011).
- Thirlwell C, Eymard M, Feber A et al. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the illumina Infinium HumanMethylation27 Bead Chip. Methods52, 248–254 (2010).
- Jasmine F, Rahaman R, Roy S et al. Interpretation of genome-wide infinium methylation data from ligated DNA in formalin-fixed, paraffin-embedded paired tumor and normal tissue. BMC Res. Notes5, 117 (2012).
- Taylor KH, Kramer RS, Davis JW et al. Ultra deep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res.67, 8511–8518 (2007).
- Korshunova Y, Maloney RK, Lakey N et al. Massively parallel bisulphite pyrosequencing reveals the molecular complexity of breast cancer-associated cytosine-methylation patterns obtained from tissue and serum DNA. Genome Res.18, 19–29 (2008).
- Suijkerbuijk KPM, van Diest PJ, van der Wall E. Improving early breast cancer detection: focus on methylation. Ann. Oncol.22, 24–29 (2011).
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl Acad. Sci. USA93, 9821–9826 (1996).
- Preusser M, Elezi L, Hainfellner JA. Reliability and reproducibility of PCR-based testing of O6-methylguanine-DNA methyltransferase gene (MGMT) promoter methylation status in formalin-fixed and paraffin-embedded neurosurgical biopsy specimens. Clin. Neuropathol.27, 388–390 (2008).
- Hamilton MG, Roldan G, Magliocco A et al. Determination of the methylation status of MGMT in different regions within glioblastoma multiforme. J. Neurooncol.102, 255–260 (2011).
- Hegi ME, Diserens AC, Gorlia T et al.MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med.352, 997–1003 (2005).
- Eads CA, Danenberg KD, Kawakami K et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucl. Acids Res.28, E32 (2000).
- Chhibber A, Schroeder BG. Single-molecule polymerase chain reaction reduces bias: application to DNA methylation analysis by bisulfite sequencing. Anal. Biochem.377, 46–54 (2008).
- Candiloro IL, Mikeska T, Hokland P, Dobrovic A. Rapid analysis of heterogeneously methylated DNA using digital methylation-sensitive high resolution melting: application to the CDKN2B (p15) gene. Epigenetics Chromatin1, 7 (2008).
- Li M, Chen WD, Papadopoulos N et al. Sensitive digital quantification of DNA methylation in clinical samples. Nat. Biotechnol.27(9), 858–863 (2009).
- Clarke J, Wu HC, Jayasinghe L et al. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol.4, 265–270 (2009).
- Flusberg BA, Webster DR, Lee JH et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods7, 461–465 (2010).
- Bock C, Lengauer T. Computational epigenetics. Bioinformatics24, 1–10 (2008).
- Bock C. Epigenetic biomarker development. Epigenomics1, 99–110 (2009).
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics18, 1427–1431 (2002).
- Tusnády GE, Simon I, Váradi A, Arányi T. BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes. Nucl. Acids Res.33, E9 (2005).
- Schuffler P, Mikeska T, Waha A, Lengauer T, Bock C. MethMarker: user-friendly design and optimization of gene-specific DNA methylation assays. Genome Biol.10, R105 (2009).
- Bock C, Reither S, Mikeska T et al. BiQ analyzer: visualization and quality control for DNA methylation data from bisulphite sequencing. Bioinformatics21, 4067–4068 (2005).
- Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucl. Acids Res.36, W170–W175 (2008).
- Lutsik P, Feuerbach L, Arand J et al. BiQ analyzer HT: locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucl. Acids Res.39, W551–W556 (2011).
- Thompson RF, Suzuki M, Lau KW, Greally JM. A pipeline for the quantitative analysis of CG dinucleotide methylation using mass spectrometry. Bioinformatics25, 2164–2170 (2009).
- Yang CH, Chuang LY, Cheng YH et al. Methyl-typing: an improved and visualized COBRA software for epigenomic studies. FEBS Lett.584, 739–744 (2010).
- Dobrovic A, Kristensen LS. DNA methylation, epimutations and cancer predisposition. Int. J. Biochem. Cell Biol.41, 34–39 (2009).
- Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res.62, 3925–3928 (2002).
- Snell C, Krypuy M, Wong EM et al.BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res.10, R12 (2008).
- Wong EM, Southey MC, Fox SB et al. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev. Res.4, 23–33 (2011).
- deVos T, Tetzner R, Model F et al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin. Chem.55, 1337–1346 (2009).
- Tanzer M, Balluff B, Distler J et al. Performance of epigenetic markers SEPT9 and ALX4 in plasma for detection of colorectal precancerous lesions. PLoS One5, E9061 (2010).
- Warren JD, Xiong W, Bunker AM et al. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. Genet.9, 133 (2011).
- Chen WD, Han ZJ, Skoletsky J et al. Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J. Natl Cancer Inst.97, 1124–1132 (2005).
- Itzkowitz S, Brand R, Jandorf L et al. A simplified, noninvasive stool DNA test for colorectal cancer detection. Am. J. Gastroenterol.103, 2862–2870 (2008).
- Zou H, Allawi H, Cao X et al. Quantification of methylated markers with a multiplex methylation-specific technology. Clin. Chem.58(2), 375–383 (2011).
- Merlo A, Herman JG, Mao L et al. 5´ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med.1, 686–692 (1995).
- Tessema M, Yu YY, Stidley CA et al. Concomitant promoter methylation of multiple genes in lung adenocarcinomas from current, former and never smokers. Carcinogenesis30, 1132–1138 (2009).
- Zhang B, Zhu W, Yang P et al. Cigarette smoking and p16 gene promoter hypermethylation in non-small cell lung carcinoma patients: a meta-analysis. PLoS One6, E28882 (2011).
- Palmisano WA, Divine KK, Saccomanno G et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res.60, 5954–5958 (2000).
- Kersting M, Friedl C, Kraus A et al. Differential frequencies of p16(INK4a) promoter hypermethylation, p53 mutation, and K-ras mutation in exfoliative material mark the development of lung cancer in symptomatic chronic smokers. J. Clin. Oncol.18, 3221–3229 (2000).
- Topaloglu O, Hoque MO, Tokumaru Y et al. Detection of promoter hypermethylation of multiple genes in the tumor and bronchoalveolar lavage of patients with lung cancer. Clin. Cancer Res.10, 2284–2288 (2004).
- Fujiwara K, Fujimoto N, Tabata M et al. Identification of epigenetic aberrant promoter methylation in serum DNA is useful for early detection of lung cancer. Clin. Cancer Res.11, 1219–1225 (2005).
- Belinsky SA, Klinge DM, Dekker JD et al. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin. Cancer Res.11, 6505–6511 (2005).
- Schmidt B, Liebenberg V, Dietrich D et al.SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer10, 600 (2010).
- Schneider KU, Dietrich D, Fleischhacker M et al. Correlation of SHOX2 gene amplification and DNA methylation in lung cancer tumors. BMC Cancer11, 102 (2011).
- Kneip C, Schmidt B, Seegebarth A et al.SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J. Thorac. Oncol.6, 1632–1638 (2011).
- Nakayama M, Bennett CJ, Hicks JL et al. Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture. Am. J. Pathol.163, 923–933 (2003).
- Ahmed H, Cappello F, Rodolico V, Vasta GR. Evidence of heavy methylation in the galectin 3 promoter in early stages of prostate adenocarcinoma: development and validation of a methylated marker for early diagnosis of prostate cancer. Transl. Oncol.2, 146–156 (2009).
- Goessl C, Krause H, Muller M et al. Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res.60, 5941–5945 (2000).
- Goessl C, Muller M, Heicappell R et al. DNA-based detection of prostate cancer in urine after prostatic massage. Urology58, 335–338 (2001).
- Nimmrich I, Sieuwerts AM, Meijer-van Gelder ME et al. DNA hypermethylation of PITX2 is a marker of poor prognosis in untreated lymph node-negative hormone receptor-positive breast cancer patients. Breast Cancer Res. Treat.111, 429–437 (2008).
- Maier S, Nimmrich I, Koenig T et al. DNA-methylation of the homeodomain transcription factor PITX2 reliably predicts risk of distant disease recurrence in tamoxifen-treated, node-negative breast cancer patients – technical and clinical validation in a multi-centre setting in collaboration with the European Organisation for Research and Treatment of Cancer (EORTC) PathoBiology group. Eur. J. Cancer43, 1679–1686 (2007).
- Harbeck N, Nimmrich I, Hartmann A et al. Multicenter study using paraffin-embedded tumor tissue testing PITX2 DNA methylation as a marker for outcome prediction in tamoxifen-treated, node-negative breast cancer patients. J. Clin. Oncol.26, 5036–5042 (2008).
- Hartmann O, Spyratos F, Harbeck N et al. DNA methylation markers predict outcome in node-positive, estrogen receptor-positive breast cancer with adjuvant anthracycline-based chemotherapy. Clin. Cancer Res.15, 315–323 (2009).
- Weiss G, Cottrell S, Distler J et al. DNA methylation of the PITX2 gene promoter region is a strong independent prognostic marker of biochemical recurrence in patients with prostate cancer after radical prostatectomy. J. Urol.181, 1678–1685 (2009).
- Banez LL, Sun L, van Leenders GJ et al. Multicenter clinical validation of PITX2 methylation as a prostate specific antigen recurrence predictor in patients with post-radical prostatectomy prostate cancer. J. Urol.184, 149–156 (2010).
- Brock MV, Hooker CM, Ota-Machida E et al. DNA methylation markers and early recurrence in stage I lung cancer. N. Engl. J. Med.358, 1118–1128 (2008).
- Juergens RA, Wrangle J, Vendetti FP et al. Combination epigenetic therapy has efficiacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov.1, 598–607 (2011).
- Paige AJW, Brown R. Pharmaco(epi)genomics in ovarian cancer. Pharmacogenomics9, 1825–1834 (2008).
- Brandes JC, Carraway H, Herman JG. Optimal primer design using the novel primer design program: MSPprimer provides accurate methylation analysis of the ATM promoter. Oncogene26, 6229–6237 (2007).
- Esteller M, Garcia-Foncillas J, Andion E et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med.343, 1350–1354 (2000).
- Esteller M, Gaidano G, Goodman SN et al. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J. Natl Cancer Inst.94, 26–32 (2002).
- Bei R, Marzocchella L, Turriziani M. The use of temozolomide for the treatment of malignant tumors: clinical evidence and molecular mechanisms of action. Recent Pat. Anticancer Drug Discov.5, 172–187 (2010).
- Mikeska T, Bock C, El-Maarri O et al. Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J. Mol. Diagn.9, 368–381 (2007).
- Vlassenbroeck I, Califice S, Diserens AC et al. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J. Mol. Diagn.10, 332–337 (2008).
- Vassella E, Vajtai I, Bandi N et al. Primer extension based quantitative polymerase chain reaction reveals consistent differences in the methylation status of the MGMT promoter in diffusely infiltrating gliomas (WHO grade II–IV) of adults. J. Neurooncol.104, 293–303 (2011).
- Toyota M, Suzuki H, Yamashita T et al. Cancer epigenomics: implications of DNA methylation in personalized cancer therapy. Cancer Sci.100, 787–791 (2009).
- Agrelo R, Cheng WH, Setien F et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc. Natl Acad. Sci. USA103, 8822–8827 (2006).
- Gagnon JF, Bernard O, Villeneuve L, Tetu B, Guillemette C. Irinotecan inactivation is modulated by epigenetic silencing of UGT1A1 in colon cancer. Clin. Cancer Res.12, 1850–1858 (2006).
- Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature434, 917–921 (2005).
- Veeck J, Ropero S, Setien F et al.BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J. Clin. Oncol.28, E563–E564 (2010).
- Taniguchi T, Tischkowitz M, Ameziane N et al. Disruption of the Fanconi anemia–BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med.9, 568–574 (2003).
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature474, 609–615 (2011).
- Satoh A, Toyota M, Itoh F et al. Epigenetic inactivation of CHFR and sensitivity to microtubule inhibitors in gastric cancer. Cancer Res.63, 8606–8613 (2003).
- Koga Y, Kitajima Y, Miyoshi A et al. The significance of aberrant CHFR methylation for clinical response to microtubule inhibitors in gastric cancer. J. Gastroenterol.41, 133–139 (2006).
- Banno K, Yanokura M, Kawaguchi M et al. Epigenetic inactivation of the CHFR gene in cervical cancer contributes to sensitivity to taxanes. Int. J. Oncol.31, 713–720 (2007).
- Wang X, Yang Y, Xu C et al.CHFR suppression by hypermethylation sensitizes endometrial cancer cells to paclitaxel. Int. J. Gynecol. Cancer21, 996–1003 (2011).
- Ogi K, Toyota M, Mita H et al. Small interfering RNA-induced CHFR silencing sensitizes oral squamous cell cancer cells to microtubule inhibitors. Cancer Biol. Ther.4, 773–780 (2005).
- Yoshida K, Hamai Y, Suzuki T et al. DNA methylation of CHFR is not a predictor of the response to docetaxel and paclitaxel in advanced and recurrent gastric cancer. Anticancer Res.26, 49–54 (2006).
- Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2´-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res.60, 6039–6044 (2000).
- Zeller C, Dai W, Steele NL et al. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene doi:10.1038/onc.2011.611 (2012) (Epub ahead of print).
- Carethers JM, Chauhan DP, Fink D et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology117, 123–131 (1999).
- Ogawa T, Liggett TE, Melnikov AA et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle11(8), 1656–1663 (2012).
- Shen L, Kondo Y, Ahmed S et al. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res.67, 11335–11343 (2007).
- Ebert MPA, Tanzer M, Balluff B et al. TFAP2E–DKK4 and chemoresistance in colorectal cancer. N. Engl. J. Med.366, 44–53 (2012).
- Issa JP, Kantarjian HM. Targeting DNA methylation. Clin. Cancer Res.15, 3938–3946 (2009).
- Shen L, Kantarjian H, Guo Yi et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J. Clin. Oncol.28, 605–613 (2010).
- Yang AS, Doshi KD, Choi SW et al. DNA methylation changes after 5-aza-2´-deoxycytidine therapy in patients with leukemia. Cancer Res.66, 5495–5503 (2006).
- Stresemann C, Bokelmann I, Mahlknecht U, Lyko F. Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol. Cancer Ther.7, 2998–3005 (2008).
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet.8, 286–298 (2007).
- Bibikova M, Le J, Barnes B et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics1, 177–200 (2009).
- Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer5, 223–231 (2005).
- Everhard S, Tost J, El Abdalaoui H et al. Identification of regions correlating MGMT promoter methylation and gene expression in glioblastomas. Neuro. Oncol.11, 348–356 (2009).
- Siegmund KD, Laird PW. Analysis of complex methylation data. Methods27, 170–178 (2002).
- Duffy MJ, Napieralski R, Martens JW et al. Methylated genes as new cancer biomarkers. Eur. J. Cancer45(3), 335–346 (2009).
Websites
- Methyl Primer Express software for bisulfite sequencing primer design. http://docs.appliedbiosystems.com/pebiodocs/04370961.pdf
- R software package for statistical analysis. www.r-project.org
- Cancer Research UK. Biomarkers and Imaging Discovery and Development Project Grants. http://science.cancerresearchuk.org/funding/find-grant/all-funding-schemes/biomarkers-imaging-discovery-development-project-grants