
Abstract
In order to achieve optimal biological activity and desired pharmacokinetic profiles, a dithiocyclopeptide linker was designed for in vivo release of protein domains from a recombinant fusion protein. This novel in vivo cleavable disulfide linker, based on a dithiocyclopeptide containing a thrombin-sensitive sequence and an intramolecular disulfide bond, was inserted between transferrin and granulocyte colony-stimulating factor (G-CSF) recombinant fusion protein domains. After expression of the fusion protein, G-C-T, from HEK293 cells, thrombin treatment in vitro generated a fusion protein linked via a reversible disulfide bond that was quickly cleaved in vivo, separating the protein domains. After release from the fusion protein, free G-CSF exhibited an improved biological activity in a cell proliferation assay. Although reversible disulfide bonds are commonly used in protein chemical conjugation methods, to our knowledge this report is the first example of the construction of a recombinant fusion protein with a disulfide linkage for the release of the functional domain. This linker design can be adapted to diverse recombinant fusion proteins in which in vivo separation of protein domains is required to achieve an improved therapeutic effect and a desirable pharmacokinetic profile and biodistribution of the functional domain.
The Missing Link
Certain recombinant fusion proteins require short peptide linkers between protein domains to provide flexibility or increase stability during synthesis. For some fusion proteins, however, the inability to cleave these stable peptide linkers in order to separate and release the protein domains can impede their in vivo function. One alternative is to utilize a reversible disulfide linkage between the two protein domains, either through cysteine bonding or bifunctional linkers, but this requires chemical conjugation of the domains and often results in heterogeneous products unsuitable for therapeutic use. In this issue, Chen et al. at the University of Southern California (Los Angeles, CA) describe a novel in vivo cleavable disulfide peptide linker for fusion proteins that overcomes these issues. Their dithiocyclopeptide linker is based on the structure of somatostatin, where two cysteine residues form an intramolecular disulfide bond, resulting in a cyclopeptide. The peptide linker also was modified to contain a thrombin-sensitive sequence between the cysteine residues. While in vitro thrombin treatment of a fusion protein containing this linker results in cleavage of the dithiocyclopeptide linker, the protein domains remain attached through the disulfide bond. After administration into whole animals, in vivo reduction of the disulfide bond separates the two protein domains, allowing them to function independently. The authors demonstrated their method by joining granulocyte colony stimulating factor (G-CSF) and transferrin (Tf) protein domains using the dithiocyclopeptide linker to generate a fusion protein (G-C-T). In vitro, free G-CSF was only released from G-C-T treated with both thrombin and dithiothreitol and not from untreated G-C-T, and the former was twice as biologically active as the latter in a cell proliferation assay. After intravenously injecting mice with thrombin-treated but still—disulfide-intact G-C-T fusion protein, free G-CSF was detected in the blood after only 5 min; peak amounts were measured after 15 min, whereas none was detected in mice injected with untreated G-C-T.
See “Design of an in vivo cleavable disulfide linker in recombinant fusion proteins” on page 513.
Introduction
Recombinant fusion proteins have become an important class of molecules in numerous fields of biotechnology, including protein engineering and purification, drug targeting and delivery, and immunology. By genetically fusing two or more genes together, the resulting fusion protein can attain multiple functional properties derived from each of its components (Citation1). Successfully constructed fusion proteins require not only the desired component proteins, but also suitable linkers to connect the protein domains. Linkers in fusion proteins generally consist of stable peptide sequences, including the glycine-serine linker (GGGGS)n and α-helix—forming peptide linkers, such as A(EAAAK)nA (n = 2–5), which can provide structure flexibility, improve protein stability, or increase biological activity (Citation2–5). However, stable peptide linkers do not allow for the separation of the two fusion protein domains in vivo and have several limitations including steric hindrance between two functional domains, altered biodistribution and metabolism of the protein moieties due to interference with each other, and incorrect folding of the fusion protein (Citation6).
To overcome some of these potential pitfalls in stable peptide linkers, we designed a fusion protein linkage utilizing the reversible nature of the disulfide bond (Citation7). There are numerous examples of application of a disulfide linkage between two proteins in drug delivery, either by using cysteine amino acid residues in proteins or by employing hetero- or homobifunctional linkers (Citation8–10). This approach requires chemical conjugation of the two separate proteins, since recombinant technologies require expression of fusion proteins as one single polypeptide chain. However, the major obstacle for the chemical conjugation approach is that the composition and size of the final product can be heterogeneous, which is unacceptable for therapeutic use. In comparison, the novel recombinant fusion protein approach described in this report offers the significant advantage in the generation of a precisely constructed, homogeneous product. This cleavable dithiocyclopeptide linker contains a thrombin-sensitive sequence, as well as an intramolecular disulfide bond formed between two cysteine residues of the linker. Treatment in vitro with thrombin results in cleavage of the thrombin-sensitive sequence, while the disulfide linkage between the two domains of the recombinant fusion protein remains (). Conceivably, the disulfide linkage will be reduced in vivo, allowing for the protein domains to be separated and regain their individual characteristics such as biological activity, biodistribution, and/or metabolism. In this paper, we use a fusion protein containing granulocyte colony-stimulating factor (G-CSF) and transferrin (Tf) (Citation11) to demonstrate the successful construction and the in vivo cleavability of a dithiocyclopeptide linker between two domains of a recombinant fusion protein.
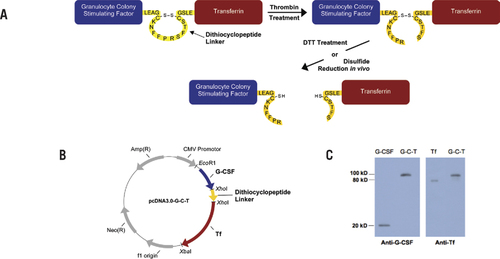
(A) Design of the recombinant fusion protein. The G-C-T fusion protein is linked via a dithiocyclopeptide linker based on the structure of somatostatin modified to contain a thrombin-specific sequence, PRS. The two cysteine residues in somatostatin naturally form a disulfide bond. (B) Plasmid encoding the G-C-T fusion protein was constructed based on the mammalian expression vector pcDNA3.0. (C) Recombinant G-C-T was analyzed by Western blot analysis using antihuman G-CSF or serum Tf primary antibody, with horseradish peroxidase—conjugated anti-rabbit or anti-goat IgG secondary antibody, respectively. The peroxidase activity was detected by enhanced chemiluminescence using the ECL substrate system.
Materials and methods
Cell lines
HEK293 cells, purchased from ATCC (Manassas, VA, USA), were grown as monolayers in DMEM with 10% (v/v) FBS at 37°C in 5% CO2. Protein-free, chemically defined CD293 medium, obtained from Invitrogen (Carlsbad, CA, USA), was used for protein expression after transfection of HEK293 cells. Murine myeloblastic NFS60 cells, provided by James Ihle (St. Jude Children's Research Hospital, Memphis, TN, USA), were grown in RPMI medium 1640 (Sigma-Aldrich, St. Louis, MO, USA) with 10% (v/v) FBS and 0.1 ng/mL recombinant mouse interleukin-3 (IL-3).
Animals
Male CF-1 mice (18–20 g) from Charles River Laboratories (Kingston, NY, USA) were used for in vivo cleavability studies. The protocol of animal experiments in this study has been approved by the Institutional Animal Care and Use Committee (IACUC) at USC. The animals were handled in accordance with the “Guide for the Care and Use of Laboratory Animals” [National Institutes of Health (NIH) Publication no.85–23, revised 1985]. The animals were fed a standard laboratory rodent diet (Purina Mills, St. Louis, MO, USA) and housed on a 12-h light/dark cycle with room temperature maintained at 22° ± 3°C and relative humidity at 50% ± 20%.
Construction of G-CSF-dithiocyclopeptide-Tf (G-C-T) plasmid
G-CSF-Tffusion protein was constructed into the mammalian expression vector pcDNA3.0 (Invitrogen) as previously described (Citation11). A dipeptide linker, Leu-Glu, was introduced between G-CSF and Tf due to the cloning site XhoI. The design of dithiocyclopeptide linker was based on the structure of the cyclopeptide, somatostatin, with the replacement of amino acids 8–10, WKT, by a thrombin-specific sequence, PRS. Two cysteine residues on somatostatin, Cys-3 and Cys-14, naturally form a disulfide bond. The oligonucleotides encoding the dithiocyclopeptide linker, synthesized by Invitrogen, were inserted between G-CSF and Tf by using the XhoI cloning site. The correct sequence was confirmed by DNA sequencing.
Expression of the recombinant G-C-T fusion protein
HEK293 cells were seeded into 175-cm2 cell culture flasks (BD Biosciences, Franklin Lakes, NJ, USA) the day before transfection in order to achieve 60–80% confluence at the time of transfection. Serum-free DMEM was used during transfection. The plasmids encoding the G-C-T fusion protein were transfected into HEK293 cells by using linear polyethylenimine (Polysciences, Warrington, PA, USA) as described (Citation12). Following a 5-h transfection, CD293 medium was used for protein expression for an additional 4-day incubation period. The conditioned medium containing the G-C-T fusion protein was centrifuged at 3000× g for 10 min to remove cell debris. The medium was then concentrated by tangential flow filtration (TFF; Millipore, Billerica, MA, USA) to a final volume of 20 mL. Buffer exchange was conducted by TFF to change the expression medium to the storage buffer (50 mg/mL mannitol, 0.1 mg/mL Tween-80, 50 mM phosphate, pH 7.0). The fusion protein was stored at −80°C until use.
Characterization of the G-C-T fusion protein by Western blot analysis
To determine the presence of G-CSF and Tf domain in the fusion protein, anti-G-CSF and anti-TfWestern blot analyses were performed. Antibody against human G-CSF (ProSci, San Diego, CA, USA) was used as primary antibody, and horseradish peroxidase—conjugated anti-rabbit IgG antibody (Sigma-Aldrich) was used as secondary antibody. Antibody against human serum Tf (Sigma-Aldrich) was used as primary antibody and horseradish peroxidase—conjugated anti-goat IgG antibody (Sigma-Aldrich) was used as secondary antibody. The peroxidase activity was detected using the enhanced chemiluminescent (ECL) substrate system (GE Healthcare, Piscataway, NJ, USA).
Characterization of the dithiocyclopeptide linker by Western blot analysis
To test whether the correct dithiocyclopeptide linker formed in G-C-T fusion protein as designed, the fusion protein was first treated by thrombin in vitro. Thrombin from bovine plasma (Sigma-Aldrich) was added to the fusion protein at 10 U/mL and incubated at 20°C for 16h to cleave the dithiocyclopeptide linker. The thrombin-treated fusion protein, which has a disulfide bond linker, is denoted as G-S-S-T. The protein samples, with or without thrombin treatment, were boiled in reducing or nonreducing loading buffer. The reducing loading buffer contains 100 mM DTT to reduce the disulfide bond linkage in the fusion protein. These samples were subjected to anti-G-CSF Western blotting analysis as described in the “Materials and methods” section.
NFS-60 proliferation assay for in vitro biological activity
In order to release free G-CSF from Tf, the G-C-T fusion protein was first treated by thrombin in vitro (as described in the “Materials and methods” section) and then incubated with 25 mM DTT at 37°C for 15 min. The reduced fusion protein was then subjected to a 100-fold dilution in RPMI 1640 medium with 10% FBS to neutralize the DTT. The thrombin-and DTT-treated fusion protein, as well as the untreated fusion protein, were further diluted to the desired concentration and were used to stimulate the proliferation of the NFS-60 cells as previously described (Citation11). Briefly, NFS-60 cells were washed by RPMI 1640 medium with 10% FBS three times to wash off the IL-3 contained in the culture medium. The cells were then seeded to 96-well plates at a density of 105 cells/well. Following the addition of serially diluted fusion protein samples, the cells were incubated at 37°C in a 5% CO2 incubator for 48 h. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was subsequently performed, incubating the cells in phenol red—free RPMI medium 1640 with 1 mg/mL MTT for 4 h. The formazan crystals that formed were then dissolved in isopropanol, and the absorbance was measured at 570 nm using a GENios Plus microplate reader (Tecan, Männedorf, Switzerland).
In vivo release of free G-CSF from G-S-S-T
The G-C-T fusion protein was pretreated with thrombin as described above, and the samples were subjected to a 4-fold dilution prior to administration. G-S-S-T or G-C-T was administered intravenously to CF1 mice via the tail vein at a dose of 4 mg/kg. After administration, blood was collected from the saphenous vein as described by Hem et al. (Citation13) at the desired time points. The collected blood was mixed with heparin to prevent clotting and was subsequently centrifuged at 500× g for 20 min to remove the blood cells. The collected plasma samples from three mice were pooled together and then analyzed by nonreducing SDS-PAGE followed by anti—G-CSF Western blot analysis, as described in the “Materials and methods” section.
Results and discussion
G-C-T fusion protein was successfully constructed and expressed
In contrast to the rapid progress in recombinant fusion protein application, the rational design of linkers in fusion proteins seems to lag behind. It has become more and more evident that with the suitable design of linkers, fusion proteins can achieve optimal biological activity and preferred structures. In order to address these concerns, we produced a recombinant disulfide-linked fusion protein by insertion of a dithiocyclopeptide linker in between G-CSF and Tf protein domains, resulting in a fusion protein, G-C-T, containing both a thrombin-sensitive cleavage site and a disulfide bond (). Following transfection and concentration of G-C-T from HEK 293 cells, Western blot analysis demonstrated that the 100-kDa protein could be detected by anti-G-CSF and anti-Tf antibodies, confirming that the fusion protein contained both G-CSF and Tf domains ().
The expression level of this dithiocyclopeptide-linked fusion protein was ∼50% of that of the fusion protein without a linker, 1.2 mg/L versus 2.5 mg/L as a typical yield. However, in human growth hormone—Tf fusion protein (Citation14), we found that this dithiocyclopeptide linker had a comparable expression level to the nonlinker fusion protein (data not shown). Therefore, it is likely that the effect of the cyclopeptide linker on the expression level of a fusion protein may depend on the nature of the protein domains as observed in other peptide linkers (Citation2). In the case of G-CSF-Tf, the expression level is only slightly decreased.
The dithiocyclopeptide linker is correctly formed in G-C-T fusion protein
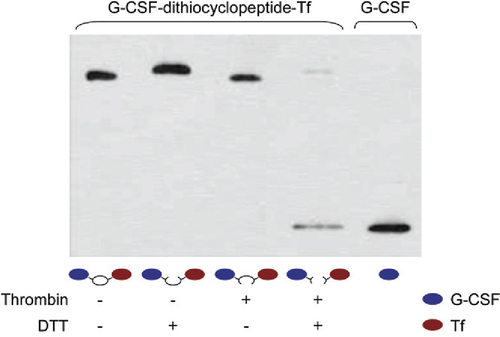
To test the correct formation of the dithiocyclopeptide linker in G-C-T, the fusion protein was treated with thrombin and/or DTT to cleave the thrombin-sensitive and disulfide linkage, respectively. As shown in , free G-CSF was only released from G-C-T after treatment with both thrombin and DTT, demonstrating the formation of both linkages. Furthermore, fusion protein aggregates were not detected in the Western blot analysis, indicating that there were only intramolecular, and not intermolecular, disulfide bonds formed. Therefore, these data show that the disulfide bond was spontaneously formed between the two cysteine residues on the linker, but not with those in the protein domains. In addition, the thrombin-sensitive sequence on the cyclic linker was recognized and cleaved by thrombin in vitro to break the stable peptide bond. By this design, we were able to create a disulfide linkage between two functional domains using recombinant fusion protein technology, a feat previously requiring the use of chemical conjugation methods of two separate proteins. We were also able to successfully generate another dithiocyclopeptide-linked fusion protein, growth hormone—Tf, which showed similar results (data not shown).
G-C-T was treated with 10 U/mL thrombin and incubated at 20°C for 16 h to generate G-S-S-T. The protein samples, with or without thrombin treatment, were boiled in reducing (containing 100 mM DTT) or nonreducing loading buffer and analyzed by anti—G-CSF Western blotting analysis as described in the “Materials and methods” section.
G-C-T has improved in vitro biological activity with the release of free G-CSF
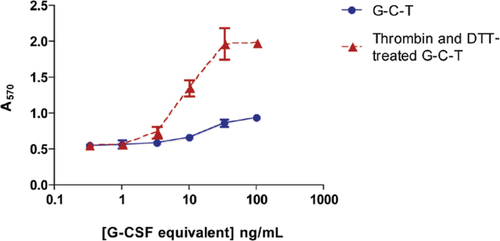
The in vitro G-CSF biological activity of the fusion protein was determined by NFS-60 cell proliferation assay (Citation11). The cells were incubated with untreated G-C-T and with G-C-T pretreated with thrombin and reduced by DTT to mimic the expected in vivo released free G-CSF. The cleaved and reduced fusion protein exhibited a 2-fold increase of in vitro biological activity, with an EC50 of 10.12 ng/mL, compared with the noncleaved, nonreduced fusion protein (i.e., EC50 = 23.69 ng/mL). In addition, the cleaved and reduced fusion protein exhibited a higher maximal response (). Therefore, the biological activity of the fusion protein was greatly improved with the release of G-CSF from Tf. It is known that the receptor binding site of G-CSF is located at both the N and C termini (Citation15). Therefore, the increased activity is likely due to the removal of Tf blockage at the C terminus following reduction. The in vitro activity results provide a proof of concept that, due to the in vivo cleavability of the linker, the fusion protein with this disulfide linker may have better in vivo biological activity than a comparable fusion protein with a noncleavable linker.
G-S-S-T was incubated with 25 mM DTT at 37°C for 15 min. The thrombin- and DTT-treated fusion protein, as well as the untreated fusion protein, were used to stimulate the proliferation of NFS-60 cells as described in the “Materials and methods” section. The DTT- and thrombin-treated fusion protein samples were subjected to a ≥100-fold dilution in RPMI 1640 medium with 10% FBS prior to cell treatment, to eliminate the effects of DTT and thrombin on the proliferation assay. An increased absorbance at 570 nm is indicative of cell proliferation.
The disulfide linkage in thrombin pretreated G-C-T is cleavable in vivo
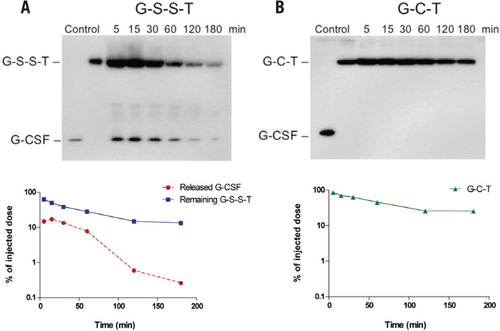
Next, we investigated the in vivo cleavage of the thrombin pretreated, disulfide-linked fusion protein (designated as G-S-S-T). G-S-S-T or G-C-T was administered intravenously to CF1 mice, and the plasma samples were analyzed by nonreducing SDS-PAGE followed by anti-G-CSF Western blot analysis. As shown in , free G-CSF could be detected in the blood as early as 5 min after injection of G-S-S-T, with a peak at ∼15 min postinjection. In contrast, no detectable amount of free G-CSF was present in the blood of CF1 mice treated with G-C-T. At the last time point of 180 min, the remaining percentage of injected dose of the G-C-T fusion protein was ∼2× higher than the G-S-S-T fusion protein. These results demonstrate that the disulfide linkage in G-S-S-T could indeed be cleaved in vivo.
(A) G-S-S-T or (B) G-C-T was administered intravenously to CF1 mice via the tail vein at a dose of 4 mg/kg. After administration, the collected blood from three mice was pooled together and analyzed by nonreducing SDS-PAGE followed by anti—G-CSF Western blot analysis as described in the “Materials and methods” section. The amount of protein in each band was quantified using Quality One software (Bio-Rad Laboratories, Hercules, CA, USA), and the percentage was calculated based on the injected dose.
The in vivo experiment in CF1 mice supported our hypothesis that the disulfide linkage created by the dithiocyclopeptide linker is cleavable in vivo. We observed a rapid release of G-CSF from the fusion protein and a quick elimination of free G-CSF due to its short in vivo half-life (). The reversible nature of the disulfide bond has been widely applied for drug delivery and drug targeting (Citation16). A well-known example for disulfide-linked conjugate is immunotoxin, in which the disulfide bond is reduced in the cytosol to release the active toxin domain. However, following administration of immunotoxin, it was found to be broken down in the circulation due to the instability of the disulfide bond in vivo (Citation17–19). The site and the mechanism of the in vivo reduction of the disulfide linkage in the fusion protein have not been fully elucidated. It has been suggested that the disulfide bond was possibly reduced in the liver. It has been demonstrated that a disulfide linked insulin-Tf conjugate could release insulin upon its incubation with rat liver slices, but not when the latter has been depleted of glutathione (Citation20). Since the liver is the major organ for glutathione production (Citation21), the higher local glutathione concentration in the liver may facilitate the reduction. In addition, a redox enzyme, protein disulfide isomerase (PDI) has been identified on hepatocyte plasma membranes in addition to its major site of localization in endoplasmic reticulum (ER) (Citation22). The membrane-associated PDI is required for the activation of diphtheria toxin by reducing the disulfide bridge between the catalytic subunit and receptor binding subunit inside the endosome (Citation23) and may have similar function to reduce the disulfide bond in our fusion protein.
In this report, we take advantage of the reversible nature of the disulfide bond to design an in vivo cleavable linker in a recombinant fusion protein to ensure the release of the active domain into the blood circulation. To our knowledge, this is the first example of the construction and characterization of a recombinant fusion protein linked through the use of a reducible disulfide bond. This linker can be utilized for a wide range of fusion proteins wherein the in vivo separation of the domains is required for achieving optimal activity or desirable biological functions.
Competing interests
The authors declare no competing interests.
Reports
Download PDF (72.1 KB)Acknowledgments
This work was supported in part by the National Institutes of Health (NIH; grant no. R01GM063647). W.C.S. is John A. Biles Professor of Pharmaceutical Sciences. This paper is subject to the NIH Public Access Policy.
References
- Uhlen, M., G.Forsberg, T.Moks, M.Hartmanis, and B.Nilsson. 1992. Fusion proteins in biotechnology. Curr. Opin. Biotechnol.3:363–369.
- Amet, N., H.F.Lee, and W.C.Shen. 2009. Insertion of the designed helical linker led to increased expression of tf-based fusion proteins. Pharm. Res.26:523–528.
- Bai, Y. and W.C.Shen. 2006. Improving the oral efficacy of recombinant granulocyte colony-stimulating factor and transferrin fusion protein by spacer optimization. Pharm. Res.23:2116–2121.
- Wriggers, W., S.Chakravarty, and P.A.Jennings. 2005. Control of protein functional dynamics by peptide linkers. Biopolymers80:736–746.
- Arai, R., H.Ueda, A.Kitayama, N.Kamiya, and T.Nagamune. 2001. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng.14:529–532.
- Yao, Z., W.Dai, J.Perry, M.W.Brechbiel, and C.Sung. 2004. Effect of albumin fusion on the biodistribution of interleukin-2. Cancer Immunol. Immunother.53:404–410.
- Shen, W.C. 1999. Emerging targeting concepts membrane-associated protein thiol-disulfide interchange activity: a potential target for antiviral and anti-tumor drug design. J. Drug Target.6:387–389.
- Shah, D. and W.C.Shen. 1996. Transcellular delivery of an insulintransferrin conjugate in enterocyte-like Caco-2 cells. J. Pharm. Sci.85:1306–1311.
- Widera, A.S., K.-J.Kim, E.D.Crandall, and W.-C.Shen. 2003. Transcytosis of GCSF-transferrin across rat alveolar epithelial cell monolayers. Pharm. Res.20:1231–1238.
- Widera, A., Y.Bai, and W.C.Shen. 2004. The transepithelial transport of a G-CSF-transferrin conjugate in caco-2 cells and its myelopoietic effect in BDF1 mice. Pharm. Res.21:278–284.
- Bai, Y., D.K.Ann, and W.C.Shen. 2005. Recombinant granulocyte colony-stimulating factor-transferrin fusion protein as an oral myelopoietic agent. Proc. Natl. Acad. Sci. USA102:7292–7296.
- Aricescu, A.R., W.Lu, and E.Y.Jones. 2006. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr.62:1243–1250.
- Hem, A., A.J.Smith, and P.Solberg. 1998. Saphenous vein puncture for blood sampling of the mouse, rat, hamster, gerbil, guinea pig, ferret and mink. Lab. Anim.32:364–368.
- Amet, N., W.Wang, and W.C.Shen. 2010. Human growth hormonetransferrin fusion protein for oral delivery in hypophysectomized rats. J. Control. Release141:177–182.
- Tamada, T., E.Honjo, Y.Maeda, T.Okamoto, M.Ishibashi, M.Tokunaga, and R.Kuroki. 2006. Homodimeric cross-over structure of the human granulocyte colony-stimulating factor (GCSF) receptor signaling complex. Proc. Natl. Acad. Sci. USA103:3135–3140.
- Saito, G., J.A.Swanson, and K.D.Lee. 2003. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Deliv. Rev.55:199–215.
- Thorpe, P.E., P.M.Wallace, P.P.Knowles, M.G.Relf, A.N.Brown, G.J.Watson, R.E.Knyba, E.J.Wawrzynczak, and D.C.Blakey. 1987. New coupling agents for the synthesis of immunotoxins containing a hindered disulfide bond with improved stability in vivo. Cancer Res.47:5924–5931.
- Letvin, N.L., V.S.Goldmacher, J.Ritz, J.M.Yetz, S.F.Schlossman, and J.M.Lambert. 1986. In vivo administration of lymphocytespecific monoclonal antibodies in nonhuman primates. In vivo stability of disulfide-linked immunotoxin conjugates. J. Clin. Invest.77:977–984.
- Arpicco, S., F.Dosio, P.Brusa, P.Crosasso, and L.Cattel. 1997. New coupling reagents for the preparation of disulfide cross-linked conjugates with increased stability. Bioconjug. Chem.8:327–337.
- Xia, C.Q., J.Wang, and W.C.Shen. 2000. Hypoglycemic effect of insulin-transferrin conjugate in streptozotocin-induced diabetic rats. J. Pharmacol. Exp. Ther.295:594–600.
- DeLeve, L.D. and N.Kaplowitz. 1991. Glutathione metabolism and its role in hepatotoxicity. Pharmacol. Ther.52:287–305.
- Noiva, R. 1999. Protein disulfide isomerase: the multifunctional redox chaperone of the endoplasmic reticulum. Semin. Cell Dev. Biol.10:481–493.
- Mandel, R., H.J.Ryser, F.Ghani, M.Wu, and D.Peak. 1993. Inhibition of a reductive function of the plasma membrane by bacitracin and antibodies against protein disulfide-isomerase. Proc. Natl. Acad. Sci. USA90:4112–4116.

