
Abstract
Clinical approaches for tumor treatment often rely on combination therapy where a DNA damaging agent is used in combination with a DNA repair protein inhibitor. For this reason, great efforts have been made during the last decade to identify inhibitors of DNA repair proteins or, alternatively, small molecules that specifically alter protein stability or trafficking. Unfortunately, when studying these drug candidates, classical biochemical approaches are prone to artifacts. The apurinic/apyrimidinic endonuclease (APE1) protein is an essential component of the base excision repair (BER) pathway that is responsible for repairing DNA damage caused by oxidative and alkylating agents. In this work, we combined conditional gene expression knockdown of APE1 protein by RNA interference (RNAi) technology with re-expression of an ectopic recombinant form of APE1 fused with the photoconvertible fluorescent protein (PCFP) Dendra2. Dendra2 did not alter the subcellular localization or endonuclease activity of APE1. We calculated APE1 half-life and compared these results with the classical biochemical approach, which is based on cycloheximide (CHX) treatment. In conclusion, we combined RNAi and in vivo confocal microscopy to study a DNA repair protein demonstrating the feasibility and the advantage of this approach for the study of the cellular dynamic of a DNA repair protein.
Chasing protein turnover
When talking about protein interactions, how long a protein remains active in the cell is an important part of the story. Protein half-lives are often determined by pulse-chase experiments with radiolabeling or by treating cells with the chemical cycloheximide (CHX) to inhibit protein synthesis, followed by immunoblotting. While CHX treatment is less laborious and lower in cost than pulse-chase, it is also toxic to cells and lacks specificity, indiscriminately inhibiting synthesis of all transcripts. Neither method allows measurement of proteins in single cells, experiments where fluorescently labeled proteins are ideal. With fluorescence loss in photobleaching (FLIP) or fluorescence recovery after photobleaching (FRAP) approaches, fluorescently tagged proteins can be photobleached, allowing researchers to monitor fluorescence restoration in the bleached area. These techniques offer information on protein trafficking and diffusion, but tagged proteins are continuously made and degraded, so measuring protein turnover and temporal expression of GFP-labeled proteins is often impossible. Moreover, in many instances, GFP fusion proteins are expressed in addition to the endogenous native form of the protein, resulting in overexpression that further complicates protein turnover measurements. In the current issue of BioTechniques, Tell et al. from the University of Udine (Udine, Italy) detail their development of a method to overcome such drawbacks by using the photoconvertible fluorescent protein (PCFP) Dendra2, which changes from green to red fluorescence in response to ultraviolet or blue light irradiation. Using Dendra2 fused to APE1, an endonuclease important for base excision repair, in combination with RNAi-mediated knockdown of endogenous APE1, the authors were able to successfully determine the APE1 half-life at physiological levels. Red and green fluorescence signals were quantified before and after photoconversion with confocal microscopy and then used to calculate the rate of APE1 turnover. This new approach proved to be more accurate than CHX methods, avoided artifacts that readily occur with immunolabeling approaches, and should be adaptable to a wide range of proteins in the future.
See “Combining RNAi and in vivo confocal microscopy analysis of the photoconvertible fluorescent protein Dendra2 to study a DNA repair protein” on page 198.
Here we combined a specific conditional gene expression knockdown of APE1 protein by RNA interference (RNAi) technology with the re-expression of an ectopic recombinant form of APE1 in fusion with the photoconvertible fluorophore (PCFP) Dendra2. Through this approach, we were able to (i) study the protein dynamics of APE1 in a physiological rather than over expression system, (ii) discriminate between protein newly synthesized or already present within the cell, and (iii) calculate APE1 half-life.
Investigation of protein dynamics is essential for understanding cellular regulation. Visualization of protein subcellular distribution and co-localization with other proteins is generally achieved using labled antibodies. Antibodybased techniques require fixation and permeabilization of the cell in order to guarantee reagents access to all subcellular compartments. Although fluorescence microscopy approaches are routinely used, the question of how well protein localization is preserved in cells after fixation and immunostaining procedures is still a matter of debate. In a recent paper, Schnell et al. highlighted how fixation and permeabilization procedures can affect epitope accessibility and therefore may create artifacts (Citation1).
An alternative approach is to express a recombinant version of the protein of interest fused to a fluorescent protein (FP), a technique that revolutionized cell biology by allowing visualization and tracking of proteins in real time at high spatio-temporal resolution (Citation2). However, this approach can also lead to erroneous results because fusing the FP with the protein of interest may alter its folding, localization or interaction with other proteins or nucleic acids, thereby affecting its function. Furthermore, the ectopic fusion protein is expressed in addition to the endogenous native protein, resulting in overexpression of the protein of interest.
The dynamic and kinetic behavior of a fluorescent fusion protein is typically determined using techniques such as FRAP or FLIP (Citation5). In both cases, a small patch of the viewable area of the cell receives high intensity illumination, which causes the fluorescence lifetime of the FP to quickly elapse. Destroying the FP and watching the repopulation into the bleached area can reveal information about organelle continuity, protein trafficking, diffusion coefficient, and mobile fraction of a protein (Citation6). However, GFP-tagged proteins are continuously synthesized, folded, and degraded within the cell (Citation2), thus rendering the measurement of protein turnover, the analysis of the temporal expression pattern, and the assesment of protein function difficult, if not impossible. Therefore, to extract information on protein degradation or movement within a cell, one would need to block the synthesis of new GFP containing molecules. To overcome this problem, Terskikh et al. designed a FP with an emission spectrum that changes with time. This type of FP, called a timer proteins or a fluorescent timer (FTs), allows estimation of the age of fusion proteins by measuring the ratio of green and red fluorescence (Citation7). FTs permit spatial and temporal observation of protein behavior in living cells.
Protein degradation is an important aspect of cellular physiology, helping to maintain the proper concentration of a protein in a living cell. Protein half-life is commonly determined by radioactive pulse-chase analysis (Citation8). This method is laborious and requires radiolabeling procedures. An alternative biochemical approach to estimate protein half-life involves time course treatment with cycloheximide (CHX) to inhibit protein synthesis, coupled with immunoblotting. CHX acts by binding the E site of the large ribosomal subunit, thus blocking elongation (Citation9). The main advantages of this technique are the reversibility of CHX treatment and the low cost. However, CHX treatment indiscriminately inhibits protein biosynthesis of all primary transcripts present within the cell and has been reported to induce apoptosis in human cell lines (Citation10). The absence of selectivity toward a specific protein and cellular toxicity are the main limitations of this method for the determination of protein half-life. An alternative fluorescence microscopy-based approach for studying protein lifetimes uses photoactivatable fluorescent proteins (PAFPs) that display a steady state minimal fluorescence that is increased by radiation with a specific wavelength, thereby allowing monitoring of turnover and degradation of the fusion protein (Citation11). In addition, neither pulse-chase nor CHX treatment allows real-time measurements at the single cell level.
The state of the art for investigating protein dynamics and lifetime relies on photoconvertible fluorescent proteins (PCFPs). This class of fluorescent molecules undergoes a switch (photoconversion) in its spectral emission properties in response to UV-violet (350–420 nm) or blue light (e.g., 488 nm) irradiation. Kaede, mKikGR, mEos2, and Dendra2 are the most frequently used PCFPs; they mature as green-fluorescent molecules and are irreversibly photoconverted to a stable brightred fluorescent state that can be tracked for several hours without photobleaching (Citation12,Citation13).
Dendra2 is a human codon-optimized variant of the octocoral Dendronephthya sp. fluorescent protein, Dendra, which has been engineered for faster maturation and brighter fluorescence both before and after photoconversion. Dendra2 matures efficiently at both 20°C and 37°C, and undergoes irreversible photoconversion from green to red in response to intense irradiation at both 405 nm and 488 nm. While non-photoconverted (green) Dendra2 possesses excitation/emission maxima at 409/507 nm, photoconverted (red) Dendra2 possesses excitation/emission maxima at 553/573 nm. Neither toxicity nor photobleaching was reported using Dendra2 (Citation12). Here we propose the use of Dendra2 activation within a whole cell to monitor APE1/Ref-1 protein degradation.
Apurinic apyrimidinic endonuclease/redox effector factor 1 (APE1/Ref-1; henceforth referred to as APE1) is a master regulator of cellular response to oxidative stress and plays a central role in the maintenance of the genome stability (Citation14). Indeed, APE1 is the major human apurinic/apyrimidinic (AP) endonuclease, exerting a pivotal role in the base excision repair (BER) pathway. APE1 is able to recognize an AP site and cleave the phosphodiester bonds 5′ to the AP site, leaving a 3′-hydroxyl group and a 5′-deoxyribose phosphate (dRP) termini flanking the nucleotide gap (Citation15). The C terminus exerts the enzymatic activity on the abasic sites of DNA while the N terminus, containing the nuclear localization signal sequence, is principally devoted to the redox transcriptional co-activation activity (Citation16). Through its redox domain, APE1 can act as a redox-regulatory protein to maintain several transcription factors (Egr-1, NF-κB, p53, HIF-1α, AP-1, and Pax proteins) in an active reduced state (Citation17). Interestingly, this protein is able to repress its own transcription (Citation18).
APE1 is a multifunctional protein and its physiological relevance is underscored by the fact that nullizygous mice for APE1 show early stage embryonic lethality (Citation14). Moreover, down-regulation of APE1 expression levels in human cells through RNAi leads to AP site accumulation, reduced cell proliferation, and triggering of apoptosis (Citation19). Increased expression of APE1 is associated with several tumorigenic processes (Citation20). As a fundamental component of the BER pathway, APE1 is an intriguing candidate for novel therapeutic strategies aimed at inhibiting DNA repair as an effective adjuvant treatment in cancer therapy (Citation21).
Under normal conditions, APE1 mainly localizes within the nuclear compartment and accumulates in the nucleoli, but it was also shown to localize in mitochondria where it is involved in mtBER activity (Citation22). Importantly, a higher intracellular expression and a robust cytoplasmic localization of APE1 have been described in lung, ovarian, thyroid, and breast cancer to correlate with higher tumor aggressiveness (Citation20). The possible causal role played by this particular distribution in tumor progression is, however, unknown. It is therefore important to investigate the mechanisms regulating APE1 localization and subcellular distribution and to acquire more accurate information about its protein synthesis rate and lifetime.
Given the many fundamental functions played by APE1 overexpression, as well as silencing, strategies to study these issues may give rise to artifacts. For this reason, we generated and characterized a cellular model introducing APE1 expressed as a fusion protein with Dendra2 in APE1-silenced cell clones, thus maintaining physiological expression levels of APE1 protein. We confirmed that the presence of Dendra2 expressed in fusion with APE1 did not alter its subcellular localization nor its enzymatic activity on abasic DNA. Then we used this cellular model to calculate APE1 half-life. Dendra2 has been used previously to follow the degradation of target proteins (Citation23), but our approach is the first study combining RNAi knock down (KD) with the re-expression of a PCFP fusion protein. In this study, we focused our attention on a DNA repair protein, but our approach is potentially applicable to a variety of protein of interests.
Materials and methods
Plasmid preparation
For generating the APE1 KI clone, APE1 cDNA was sub-cloned from a pFLAG-CMV-5.1/APE1vector (Sigma Aldrich, Milan, Italy) into the pDendra2-N vector (Clontech, Mountain View, CA) in frame with the Dendra2 coding sequence to generate an APE1-Dendra2 fusion protein. To avoid degradation of ectopic APE1 mRNA by the specific siRNA sequence, two nucleotides (+180 and +183) of the APE1 cDNA coding sequence were mutated with site-directed mutagenesis kits (Stratagene, Milan, Italy), leaving the APE1 amino acid sequence unaffected (Citation20). To generate cell clones stably expressing APE1-Dendra2 or Dendra2 proteins, vectors were linearized before transfection by digestion with HpaI restriction enzyme (Fermentas, Milan, Italy), purified through ethanol precipitation, and finally quantified with Nanodrop (Thermo Scientific, Milan, Italy).
Cell clone generation and culture
The HeLa APE1 inducible siRNA clone was previously generated and characterized in our lab (Citation24). Briefly, cells were grown in DMEM high glucose (PAA, Milan, Italy) supplemented with 2 mM stable L-Glutamine, 10% fetal bovine serum (both from Euroclone, Milan, Italy), 100 U/mL penicillin, 10 µg/mL streptomycin sulfate, 3 µg/mL Blasticidine and 100 µg/mL Zeocine (all from Invitrogen, Monza, Italy). To induce siRNA expression, 1µg/mL doxycycline (Sigma Aldrich) was added to the medium and cells were grown for the reported amount of time. Dendra2 and APE1-Dendra2 reconstituted clones were generated by transfecting the above-mentioned siRNA clone with 2 µg of linear pDendra2-N or pDendra2-N-APE1 expressing vectors, respectively. A day before transfection, 4 × 105 cells were seeded into a 6 well plate and transfection was carried out using Lipofectamine 2000 (Invitrogen) following manufacturer instructions. After a 48 h transfection period, cells were grown in the presence of 800 µg/mL G-418 (Invitrogen) for 14 days and selected for the acquired resistance. Single clones were isolated by using cell cloning cylinders (Sigma Aldrich), and expanded to 2 × 106 cells in the presence of selective antibiotics. For calculation of APE1 half-life, CHX (Sigma Aldrich) was resuspended in DMSO and added to the cell medium at a final concentration of 100 µg/mL.
Confocal microscopy analysis
For confocal microscopy analysis of live cells, 4 × 105 cells were seeded onto glass bottom WillCo Wells Petri dishes (Ted Pella Inc., Redding, CA) and grown in DMEM without phenol red to avoid interference from medium auto-fluorescence. Images reported in and , and Supplementary Figure sS1 and S3, were obtained using a Leica TCS SP confocal microscope equipped with a temperature control unit and 405 nm diode, 458/488 nm argon and 563/683 nm HeNe lasers, RSP 500 filter, and 63×/1.32 oil CS. Acquisition parameters were 488 nm excitation line at 15% laser power, emission filter BP (band pass) 500–535 nm, PMT at 760V and 543 nm excitation line at 70% laser power, emission filter BP 555–620 nm and PMT 820V were used for channel 1 (Ch1) and channel 2 (Ch2) accordingly. Images reported in , Supplementary Figure S2, and Supplementary Movie S1 were obtained using a Zeiss LSM 710 confocal microscope equipped with a full living-cell system and 405 nm diode, 458/488/514 nm argon and 633 nm HeNe lasers, FSet 10 filter, and 63×/1.40 oil DIC M27 objective. Acquisition parameters were BP 488 nm SP mirror, BP 515–565 nm with laser power 5%, and PMT 760V in Ch1; BP 555 nm SP mirror, BP 575–640 nm with laser power 70%, and PMT 880V in Ch2. All images were collected at 1024 × 1024 pixel resolution. A 405 nm diode laser was employed at 100% power for a single scan with a pixel dwell time of 1.27 µs for total photoconversion of Dendra2. To obtain partial photo-conversion of the fluorescent protein, laser power was reduced to 30%.
(A) Inducible APE1 siRNA HeLa cell clones were stably transfected with the pDendra2-N empty vector or a vector encoding the APE1-Dendra2 fusion protein. The expression of endogenous and ectopic APE1 was evaluated by Western blotting with total cell extracts before and after expression of the specific APE1 siRNA sequence through doxycycline treatment. Assays were performed by immunoblotting with the specific anti-APE1 antibody or anti-Actin as a loading control. (B) Live confocal analysis of Dendra2 and APE1-Dendra2 clones. Dendra2 protein localizes within cytosol and nuclei but is completely excluded from nucleoli (red arrows). Expression of Dendra2 in fusion with APE1 determines the localization of the recombinant protein within the nuclear compartment and its accumulation in the nucleoli (red arrows). (C) Confocal analysis of HeLa cells expressing Dendra2 and APE1-Dendra2 proteins. After transfection, HeLa cells were analyzed in vivo or fixed with PFA 4% for 20 min. In vivo Dendra2 is excluded from the nucleoli while the fixation procedure generates an artifact showing uniform nuclear staining.
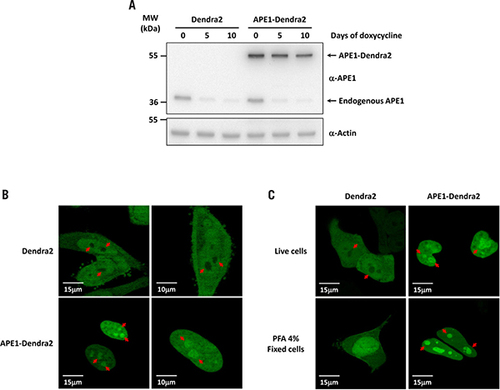
(A) Schematic representation of the enzymatic assay established to measure APE1 endonuclease activity on dsDNA. (B) Western blotting analysis of 5 µg of nuclear cell extracts from Dendra2 and APE1-Dendra2 clones treated (+) or not (-) with doxycycline for 10 days. Relative amounts of APE1 protein are reported at the bottom. (C) APE1 endonuclease activity on abasic dsDNA is rescued by the expression of the APE1-Dendra2 ectopic recombinant protein. The conversion of the radiolabeled THF-containing oligonucleotide substrate (S) to the shorter incised product (P) was evaluated on a denaturing 20% (wt/vol) polyacrylamide gel. A representative image from three independent experiments is shown. Average values of incision percentage with standard deviations of three independent experiments are reported in the graph.
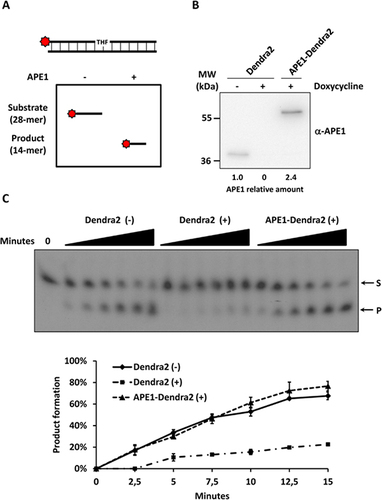
(A) Western blotting analysis of 15 µg of WCE from HeLa cells treated with CHX for the reported amount of time. Densitometric analysis of APE1 relative abundance reported in the graph identify APE1 half-life to be 9 h. Ponceau S staining of the blotted membrane was used as loading control. (B) Representative merged images of APE1-Dendra2 cells analyzed with confocal microscopy before and after Dendra2 photoconversion. (C) Each time point represents the average fluorescence and standard deviation of Dendra2 green (not-photoconverted) and red (photoconverted) calculated on 15 cells before and after UV pulse up to 14 h. The fluorescence half-life of Dendra2 after photoconversion was estimated at 7 h.
Cell extracts and Western blotting analysis
For preparation of total protein lysate, cells were harvested by trypsinization and centrifuged at 250 × g for 5 min at 4°C. Supernatant was removed, and the pellet was washed once with ice-cold PBS and then centrifuged again as described. The cell pellet was resuspended in Lysis buffer solution (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) at a density of 107 cells/mL and incubated for 30 min at 4°C. After centrifugation at 12,000 × g for 30 min at 4°C, the supernatant was collected as total cell lysate. Protein concentration was determined using Bio-Rad protein assay reagent (Bio-Rad, Milan, Italy). For preparation of nuclear enriched fractions, cells were harvested as described, and resuspended in T1 Solution (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM MgCl2, 0.1 mM EDTA pH 8.0) at a cell density of 2 × 107 cells/mL. Nuclei were isolated by centrifugation at 800 × g for 10 min at 4°C, while the supernatant was collected as cytoplasmic cell lysate. Nuclei were then washed with 500 mL of T1 solution and collected by centrifugation as described. Nuclei were subsequently lysed with T2 Solution (20 mM HEPES pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.1 mM EDTA pH 8.0, 5% glycerol), incubated for 30 min at 4°C, and centrifuged at 12,000 × g for 30 min at 4°C. The supernatant was collected as nuclear cell lysate and protein concentration was determined using Bio-Rad protein assay reagent (Bio-Rad). All cell lysis solutions were supplemented with Protease inhibitor cocktail (Sigma Aldrich), 0.5 mM PMSF, and DTT 1 mM. The reported amount of total or nuclear protein extracts was separated onto a 12% SDS-PAGE followed by Western blotting. The presence of APE1 and APE1-Dendra2 fusion protein was detected using an anti-APE1 monoclonal antibody (1:2000) (Novus, Littleton, CO), and anti-Actin rabbit polyclonal antibody (1:2000) (Sigma Aldrich) was used as a loading control.
Endonuclease assay
A 28-mer oligonucleotide containing a tetrahydrofuran (THF) residue mimicking an abasic site was labeled in vitro with 32P and annealed with its complementary sequence (Citation25). APE1 recognizes the THF abasic site and cleaves the phosphodiester backbone immediately 5′ to the AP site creating a nick. Reactions were separated in a urea 6 M denaturing 20% polyacrylamide gel, which allowed discrimination of unprocessed substrate (28-mer) and cleaved product (14-mer) by exposing the gel to autoradiography film (Sigma Aldrich).
Results and discussion
Dendra2 does not alter APE1 subcellular localization
HeLa cells were used as a cellular model to generate an inducible reconstituted knock-in (KI) clone expressing an ectopic recombinant form of APE1 protein in fusion with Dendra2 at the C terminus. Endogenous expression of APE1 was knocked-down by siRNA technology in a conditional manner through a doxycycline-responsive promoter (Citation19). Ectopic expression was achieved by stably cloning an siRNA-resistant cDNA APE1 in fusion with Dendra2 sequence (APE1-Dendra2). As a control, we used the empty vector expressing only Dendra2 (Dendra2). Inducible expression of specific APE1 siRNA sequences by doxycycline treatment efficiently promoted endogenous APE1 down-regulation in both control and KI clones. After 10 days of treatment, endogenous APE1 expression was almost undetectable (less than 10%) as compared with untreated cells, while ectopic APE1-Dendra2 fusion protein levels were slightly affected (). This time point was therefore chosen for further experiments. Comparison of APE1 protein amounts between the endogenous form in the Dendra clone at point zero and the ectopic fusion protein in the APE1-Dendra clone after 10 days of doxycycline treatment showed similar expression levels (1.8 ± 0.6 -fold).
Next, we evaluated if the presence of Dendra2 at the C terminus of APE1 affected the localization of the ectopic protein within the different subcellular compartments. Live confocal microscopy analysis of the Dendra2 clone () showed green fluorescence corresponding to Dendra2 before photoconversion in both the cytoplasmic and nuclear compartments, while nucleoli were completely excluded (red arrows). In contrast, expression of APE1 fused to Dendra2 showed green fluorescence in nuclei and the accumulation of the ectopic fusion protein within nucleolar structures (red arrows: ), results consistent with our previous work (Citation19,Citation26). Similar results were also obtained when transiently transfecting pDendra2-N and pDendra2-N-APE1 vectors into the human neuroblastoma cell line SF767 (Supplementary Figure S1). Taken together, these data lead to the conclusion that the presence of Dendra2 does not alter the subcellular localization of APE1. Next, we evaluated if a fixation protocol commonly used for immunofluorescence analysis would alter Dendra2 subcellular localization. Dendra2 displays uniform cytoplasmic/nuclear staining and does not localize to nucleoli (), but cell fixation with PFA 4% for 20 min before confocal analysis caused Dendra2 to accumulate in the nucleus and nucleolar compartment (). With the APE1-Dendra2 clone, we observed a more robust signal corresponding to nucleoli following fixation. These data confirm that mild fixation can alter protein localization (), while strong fixation ensures protein immobilization and ultrastructure preservation but may interfere with epitope recognition and penetration of antibodies (Citation1). Therefore, we conclude that studies of APE1 subcellular trafficking could be performed more reliably by adopting a live microscopy analysis rather than an immunofluorescence-based approach.
APE1-Dendra2 re-expression in APE1 knocked down cell clone rescues the loss of AP-endonuclease activity
A standard assay to measure APE1 endonuclease activity uses a dsDNA oligonucleotide containing one 32P radio labeled 5′ strand with a central tetrahydrofuran (THF) residue that mimics an AP site. APE1 is able to recognize the AP site and cleave the phosphodiester bond to generate a shorter oligonucleotide (Citation25). We quantified APE1 endonuclease activity by incubating a cell extract with such a dsDNA probe, separating the reaction mixture in a denaturing polyacrylamide gel, and exposing the gel to a radiographic film ().
In order to evaluate if the presence of the fusion fluorescence protein altered APE1 endonuclease activity, Dendra2 and APE1-Dendra2 KI clones were treated (+) or not (-) with doxycycline for 10 days, and nuclear protein extracts were isolated under native conditions. Western blotting analysis confirmed the absence of endogenous APE1 in both clones after doxycycline treatment and the presence of the ectopic recombinant Dendra2-APE1 fusion protein in the KI clone. Densitometric analysis highlighted a 2.4-fold higher amount of APE1-Dendra2 protein in the KI clone with respect to the amount of endogenous APE1 protein in the Dendra2 untreated clone (). Therefore, to compare the endonuclease activity between endogenous and ectopic recombinant APE1 100 ng nuclear extracts of Dendra2 and 42 ng of APE1-Dendra2 containing equal amounts of APE1 protein were used in the assay. The graph in shows the percentage of product formation at different time points: loss of APE1 expression led to a significant reduction (∼2.7-fold after 10 min) of AP endonuclease activity (Dendra2 +). Notably, re-expression of APE1-Dendra2 ectopic protein completely rescued APE1 enzymatic activity (). Therefore we conclude that the presence of Dendra2 in fusion at the C terminus of APE1 does not alter the endonuclease activity of the protein, thus confirming the reliability of the cellular model generated.
Dendra2 allows the calculation of APE1 protein half-life
The conventional method for calculating the half-life of a protein of interest is to inhibit protein biosynthesis by treating cells with CHX (Citation9) and then measure the reduction in expression levels of the protein of interest by Western blotting. For this purpose, we treated HeLa cells with CHX or DMSO as control at the final concentration of 100 µg/mL for up to 10 h. After the reported times, cells were harvested and 15 µg of total protein extracts were separated onto a 12% SDS-PAGE gel and analyzed by Western blotting for the presence of APE1 (). Densitometric analysis as shown in the graph represents APE1 relative levels during CHX treatment normalized to untreated cells. After 9 h, the total protein amount of APE1 decreased by 50%, but we did not observe any significant reduction of APE1 during the first 2 h of treatment. This phenomenon could be ascribed to the time required for CHX to reach an intracellular concentration sufficient to block protein neo-synthesis. In parallel, we tested the cellular model to calculate APE1 half-life in vivo by following the reduction of Dendra2 fluorescence after photoconvertion from green to red in response to violet light irradiation (Citation12). Using a 405 nm diode laser at 100% of power for a single scan at 1.27 µs pixel dwell time, it was possible to photoconvert almost all (more than 80 ± 7%) Dendra2 from green to red in a region of interest overlapping an entire cell within the field (Supplementary Figure S2). To reduce the damaging effect of irradiation, we reduced the laser power to 30% and obtained a partial photoconversion (24 ± 6%) of APE1-Dendra2 protein in the cell. In each experiment, some cells presented within the field were photoconverted while others were not irradiated and served as control. We did not observe any different behavior between these two populations in terms of cell morphology and motility, suggesting that the process of photoconversion was not harmful to the cell (Supplementary Movie S1). The absence of photobleaching, crosstalk between fluorophores, and stability of Dendra 2 have been also evaluated (data not shown). To calculate APE1 half-life after photoconversion, cells were grown in a confocal microscopy life station and imaged each hour (). Red and green fluorescence signals were quantified before and after photoconversion and then each hour for 14 consecutive hours. The graph in represents the mean fluorescence intensity of fifteen cells measured each hour in three independent experiments. The fluorescence half-life of Dendra2 after photoconversion was estimated at 7 h, about 2 h shorter than estimates obtained using CHX treatment. Similarly, APE1 half-life was estimated at 7 h in SF767 cells transiently expressing pDendra2-N-APE1 (Supplementary Figure S3). In contrast to the CHX treatment, we observed a gradual reduction of the red signal starting in the first hour of the in vivo analysis. Remarkably, this 2 h gap between the half-lives measured with the different methods fits with the delay in protein degradation during CHX treatment (). In addition, it has to be mentioned that CHX is a cytotoxic and pro-apoptotic agent that may also cause the onset of an adaptive mechanism involving the protein of interest, and therefore we cannot exclude that it could alter the half-life of APE1, a protein involved in cellular response to DNA damage and oxidative stress. In the , it is also possible to observe APE1-Dendra2 neo-synthesis. This supports our conclusion that our experimental procedures are not harmful to the cells, and therefore the half-life is accurate and not affected by experimental bias. Based on our data, we can conclude that the in vivo confocal microscopy approach is more reliable than using CHX to calculate protein life-time.
In conclusion, we generated and characterized a cellular model where endogenous APE1 was replaced by an ectopic recombinant form of the protein expressed in fusion with Dendra2. Moreover, we demonstrated that the presence of the fluorescent protein altered neither APE1 localization nor its fundamental DNA repair activity on abasic DNA. Finally, we calculated APE1 half-life using both the APE1-Dendra2 cellular model and the classical approach using the protein synthesis inhibitor CHX. This comparison provided us with a proof-of-concept that using PCFP in fusion with a DNA repair protein such as APE1 is a reliable approach for evaluating in vivo protein dynamics. It will be interesting to evaluate the practical application of this method in cancer research, where the rational combination of DNA damaging agents with DNA repair protein inhibitors offers the most promising perspective for clinical utility (Citation27), and where methodological tools are required to characterize the effects of inhibitors on protein stability, localization, and enzymatic activity.
Author contributions
G.T. and C.V. defined the research theme, designed methods and carried out the laboratory experiments, analyzed the data, interpreted the results and wrote the paper. M.D.P. and M.M.K. contributed to the microscopy analysis, data interpretation and results discussion.
Competing Interests
The authors declare no competing interests.
Additional File 1
Download Zip (11.1 MB)Acknowledgements
The authors would like to thank José Artacho and Freddy Radtke for providing technical and logistical support at the Ecole Polytechnique Fédérale de Lausanne. This work was supported by a grant from AIRC (IG10269) to G.T. and also by the Regione Friuli Venezia Giulia for the Project ‘MINA’ under the program entitled: “Programma per la Cooperazione Transfrontaliera Italia-Slovenia 2007-2013.”
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2144/000114088
Additional information
Funding
References
- Schnell, U., F.Dijk, K.A.Sjollema, and B.N.Giepmans. 2012. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods9:152–158.
- Lippincott-Schwartz, J. and G.H.Patterson. 2003. Development and use of fluorescent protein markers in living cells. Science300:87–91.
- Cubitt, A.B., R.Heim, S.R.Adams, A.E.Boyd, L.A.Gross, and R.Y.Tsien. 1995. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci20:448–455.
- Day, R.N. and M.W.Davidson. 2009. The fluorescent protein palette: tools for cellular imaging. Chem. Soc. Rev38:2887–2921.
- Lippincott-Schwartz, J., N.Altan-Bonnet, and G.H.Patterson. 2003. Photobleaching and photoactivation: following protein dynamics in living cells. Nat. Cell Biol10(Suppl):S7–S14.
- Cole, N.B., C.L.Smith, N.Sciaky, M.Terasaki, M.Edidin, and J.Lippincott-Schwartz. 1996. Diffusional mobility of Golgi proteins in membranes of living cells. Science273:797–801.
- Terskikh, A., A.Fradkov, G.Ermakova, A.Zaraisky, P.Tan, A.V.Kajava, X.Zhao, S.Lukyanov, et al.. 2000. “Fluorescent timer”: protein that changes color with time. Science290:1585–1588.
- Zhou, P. 2004. Determining protein half-lives. Methods Mol. Biol284:67–77.
- Schneider-Poetsch, T., J.Ju, D.E.Eyler, Y.Dang, S.Bhat, W.C.Merrick, R.Green, B.Shen, and J.O.Liu. 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol6:209–217.
- Martin, S.J., S.V.Lennon, A.M.Bonham, and T.G.Cotter. 1990. Induction of apoptosis (programmed cell death) in human leukemic HL-60 cells by inhibition of RNA or protein synthesis. J. Immunol145:1859–1867.
- Patterson, G.H. and J.Lippincott-Schwartz. 2002. A photoactivatable GFP for selective photolabeling of proteins and cells. Science297:1873–1877.
- Gurskaya, N.G., V.V.Verkhusha, A.S.Shcheglov, D.B.Staroverov, T.V.Chepurnykh, A.F.Fradkov, S.Lukyanov, and K.A.Lukyanov. 2006. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol24:461–465.
- Baker, S.M., R.W.Buckheit, 3rd, and M.M.Falk. 2010. Green-to-red photoconvertible fluorescent proteins: tracking cell and protein dynamics on standard wide-field mercury arc-based microscopes. BMC Cell Biol11:15.
- Izumi, T., D.B.Brown, C.V.Naidu, K.K.Bhakat, M.A.Macinnes, H.Saito, D.J.Chen, and S.Mitra. 2005. Two essential but distinct functions of the mammalian abasic endonuclease. Proc. Natl. Acad. Sci. USA102:5739–5743.
- Myles, G.M. and A.Sancar. 1989. DNA repair. Chem. Res. Toxicol2:197–226.
- Xanthoudakis, S., G.G.Miao, and T.Curran. 1994. The redox and DNA-repair activities of Ref-1 are encoded by non overlapping domains. Proc. Natl. Acad. Sci. USA91:23–27.
- Tell, G., F.Quadrifoglio, C.Tiribelli, and M.R.Kelley. 2009. The many functions of APE1/Ref-1: not only a DNA-repair enzyme. Antioxid. Redox Signal11:601–620.
- Kuninger, D.T., T.Izumi, J.Papaconstantinou, and S.Mitra. 2002. Human AP-endonuclease 1 and hnRNP-L interact with a nCaRE-like repressor element in the AP-endonuclease 1 promoter. Nucleic Acids Res.30:823–829.
- Vascotto, C., L.Cesaratto, L.A.Zeef, M.Deganuto, C.D’Ambrosio, A.Scaloni, M.Romanello, G.Damante, et al.. 2009. Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics9:1058–1074.
- Tell, G., G.Damante, D.Caldwell, and M.R.Kelley. 2005. The intracellular localization of APE1/Ref-1: more than a passive phenomenon?Antioxid. Redox Signal7:367–384.
- Tell, G., D.Fantini, and F.Quadrifoglio. 2010. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell. Mol. Life Sci67:3589–3608.
- Stuart, J.A., K.Hashiguchi, D.M.Wilson, 3rd, W.C.Copeland, N.C.Souza-Pinto, and V.A.Bohr. 2004. DNA base excision repair activities and pathway function in mitochondrial and cellular lysates from cells lacking mitochondrial DNA. Nucleic Acids Res.32:2181–2192.
- Zhang, L., N.G.Gurskaya, E.M.Merzlyak, D.B.Staroverov, N.N.Mudrik, O.N.Samarkina, L.M.Vinokurov, S.Lukyanov, et al.. 2007. Method for real-time monitoring of protein degradation at the single cell level. Biotechniques42:446–450.
- Vascotto, C., L.Cesaratto, L.A.Zeef, M.Deganuto, C.D’Ambrosio, A.Scaloni, M.Romanello, G.Damante, et al.. 2009. Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics9:1058–1074.
- Kreklau, E.L., M.Limp-Foster, N.Liu, Y.Xu, M.R.Kelley, and L.C.Erickson. 2001. A novel fluorometric oligonucleotide assay to measure O(6)-methylguanine DNA methyltransferase, methylpurine DNA glycosylase, 8-oxoguanine DNA glycosylase and abasic endonuclease activities: DNA repair status in human breast carcinoma cells overexpressing methylpurine DNA glycosylase. Nucleic Acids Res.29:2558–2566.
- Vascotto, C., E.Bisetto, M.Li, L.A.Zeef, C.D’Ambrosio, R.Domenis, M.Comelli, D.Delneri, et al.. 2011. Knock-in reconstitution studies reveal an unexpected role of Cys-65 in regulating APE1/Ref-1 subcellular trafficking and function. Mol. Biol. Cell22:3887–3901.
- Vascotto, C. and M.L.Fishel. 2012. Blockade of Base Excision Repair: Inhibition of Small Lesions Results in Big Consequences to Cancer Cells, p. 29–53. In Mark R.Kelley(Ed.), DNA Repair in Cancer Therapy: Molecular Targets and Clinical Applications. Academic Press Elsevier Inc., San Diego, CA.

