
Abstract
Manipulating gene expression in mammalian cell lines is one of the most widely used methods for studying gene function. Tetracycline- and doxycycline-inducible systems are sensitive, reproducible, relatively inexpensive, and proven to work well in both cell lines and mouse models. However, obtaining homogeneous transgene expression or uniform knockdown by short hairpin RNA requires time-consuming and labor-intensive single-cell cloning to derive stable cell lines. For this reason, Tet-inducible cell systems have yet to be widely adopted. Here we describe the XT-cell method, a novel system for establishing isogenic inducible cell lines using founder reporter lines and recombinase-mediated cassette exchange. We demonstrate that, using this XT-cell method, isogenic stable Tet-inducible cell lines can be efficiently created with much less effort and time as compared with conventional methods. The XT-plasmids and the XT-founder cell lines will be a valuable resource to researchers interested in versatile modulation of gene expression in cell culture systems, and this method has the potential to expedite many aspects of biomedical research.
The XT-cell method is a novel system for establishing isogenic inducible cell lines using founder reporter lines and recombinase-mediated cassette exchange. This method allows quick and efficient creation of Tet-inducible cell lines, saving time and labor compared with conventional methods.
Introduction
Manipulating gene expression in mammalian cell lines has been widely used to study gene functions. Conventionally, experimental manipulation of gene expression in mammalian cell lines is performed by introducing cDNA encoding the gene of interest for overexpression or by introducing short hairpin RNA or siRNA for down-regulation. Compared with transient transfection or viral transduction, single-cell-derived stable cell lines offer the advantages of relatively homogeneous transgene expression or uniform knockdown by short hairpin RNA. Moreover, stable lines can generate a large quantity of cells. The availability of inducible stable cell lines is particularly useful for functional studies of a specific gene and for drug discovery due to on-demand tunable expression.
Tetracycline- and doxycycline-inducible systems (Tet-inducible cell lines) are sensitive, reproducible, relatively inexpensive, and proven to work well both in cell lines and mouse models. However, when a DNA cassette containing the Tet-responsive promoter and transgene is randomly integrated into the genome from a plasmid or viral vector, transgene expression is influenced by the surrounding host chromatin (Citation1). These position effects can occasionally be advantageous; for example, polyclonal populations of cells with various transgene expression levels could be isolated from a pool of lentivirus-transduced cells (Citation2). However,it is generally accepted that position effects cause “leakage” or poor Tet-inducibiliy of the Tet-system in cell lines.
The conventional method for establishing homogeneous Tet-inducible cell lines depends on single-cell cloning, which is time-consuming and labor-intensive. Alternative approaches have been reported for identifying a genomic locus that potentially allows for high-inducibility (e.g., a silent but activatable (s/a) genomic locus), followed by inserting a DNA cassette into this locus using recombinase-mediated cassette exchange (RMCE) (Citation3–5). RMCE has been shown to effectively swap DNA between a genomic locus and a donor plasmid (Citation6). The DNA cassette contains the target sequence of interest (SOI) as well as the Tet-responsive promoter, which drives Tet-regulated transcription (Citation3–5). Detailed procedures for selecting cells with the preferred s/a locus have not been standardized, and the success rate of RMCE varied.
Two previous studies required an additional single-cell cloning step after RMCE to obtain homogeneous cell populations, since the correct recombination rates were shown to occur in 50%–80% of cells (Citation3, Citation4). Although a solution has been proposed to achieve a higher RMCE rate with the second single-cell cloning step omitted, founder cell lines are not selected based on tetracycline inducibility (Citation5). As a result, the efficiencies of gene knockdown vary among different founder lines (Citation5). One way to overcome this problem is to screen for multiple founder lines with acute induction and low leakage. However, the tetracycline inducibility of the founder cell lines could not be guaranteed prior to the RMCE procedure.
To circumvent the limitations of RMCE-based inducible expression systems, we created a unique plasmid set and optimized procedures to efficiently establish Tet-inducible lines. Our strategy was inspired by a routine practice in mouse knock-in and transgenic models where reporters, recombinases, or other genetic elements are introduced into the mouse genome and found to be regulated in the same manner as the endogenous gene by transcriptional regulatory elements such as promoters and enhancers (Citation7–9).
Given that transgenes such as LacZ or Cre-recombinase can faithfully recapitulate the promoter activity of the gene they replace in mouse models, we reasoned that the selection of cells in which the Tet-promoter is integrated in a locus resulting in potent inducibility and minimum leakage (founder cells) can be aided by a reporter gene encoding a fluorescent protein. The reporter gene can then be replaced using RMCE with a SOI to derive Tet-inducible cell lines in which the Tet-promoter drives the expression of the SOI in the same manner as in the founder cells.
As the first step in RMCE, a founder cell line was established to harbor a DNA cassette flanked with two incompatible recombinase recognition sites, LoxP and FRT sites, recognized by Cre and Flp recombinases, respectively (therefore named XT-cells). The second step involves introducing a cassette containing the DNA SOI and the same set of recombinase recognition sites (donor vector), which exchanges with the genomic DNA sequence through site-specific recombination with the help of the Cre and Flp recombinases. We designed a unique plasmid set (the XT-plasmid series) and demonstrated that our methodology (the XT-cell method) allows quick and efficient creation of inducible cell lines.
Materials and methods
Plasmid constructs
For establishing founder XT cell lines, we constructed two lentiviral targeting vectors: pLentiXT1 mCherry and pLentiXT2 mCherry (), using PCR and cloning methods. The backbones for the targeting vectors, CMV/TO, WPRE, and Puro, were obtained from pLenti-CMV/TO Puro DEST (Addgene plasmid 17293, Addgene, Cambridge, MA) (Citation10). The mCherry sequence was from H2B-mCherry (Addgene plasmid 20972) (Citation11). The hygro mycin resistance gene was from MSCV Luciferase PGK Hygro (Addgene plasmid 18782). The TRE-minCMV promoter and rtTA3 reverse tetracycline transactivator sequences were cloned from a pTRIPZ vector (Thermo Fisher Scientific, Pittsburgh, PA).
(A) Diagram of plasmid vectors used in this study. (B) The steps of the XT-method. Hygromycin-resistant and puromycin-sensitive XT founder reporter cell lines that show optimal tetracycline induction were established in the first step. A donor vector including the sequence of interest (SOI) was transfected with pFIC (expressing Flp and Cre recombinases) into the XT reporter cells and selected by puromycin. The resulting puromycin resistant cells are due to successful recombinase-mediated cassette exchange (RMCE) and retain tetracycline induction capability as the XT founder reporter cells.
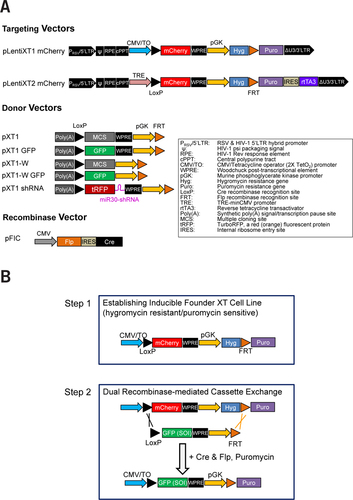
The targeting vector pLentiXT1 mCherry, based on the T-REx system (Life Technologies, Grand Island, NY), contains the CMV/TO promoter, which requires tetracycline repressor (TR) to repress CMV promoter activity. TR was introduced using pLenti6/TR (Life Technologies). The targeting vector pLentiXT2 mCherry, based on the Tet-On system (Clontech, Mountain View, CA) can be used as a single vector to establish founder XT cells, as it contains the rtTA3 gene, which can activate the TRE-minCMV promoter in the presence of doxycycline.
The donor vector pXT1 series () was constructed using pGL3.basic (Promega, Madison, WI) as backbone, and were used as the donors in RMCE. The synthetic poly(A) signal/transcription pause site in these donor vectors was included to reduce expression of the transgene during transfection. The multi-cloning site cassette was cloned from pcDNA6 V5/His.b (Life Technologies). The miR30-adapted shRNA design (TurboRFP plus shRNAmir) was adapted from the pTRIPZ vector (Thermo Fisher Scientific). The Cre and Flp (the Flpe variant) recombinases were delivered using the pFIC plasmid (Citation12). ARID1A cDNA and shRNA (CCTCTCTTATACACAGCAGAT) were described previously (Citation13). Because the woodchuck post-transcriptional element or post-transcriptional regulatory element (WPRE) can stabilize mRNA and increase transgene expression (Citation14), the pXT1 series included one with the WPRE sequence (pXT1), and one without the WPRE sequence (pXT1-W), which can be chosen based on the desired expression level.
Establishing founder XT cells
Before using pLentiXT1 mCherry to establish founder XT reporter cells, pLenti6/TR was first transduced into cells, and cells were selected in Blasticidin (Life Technologies) as pooled clones (HeLa and HEC-1-A cells). The pLentiXT2 mCherry vector was used as a single agent to establish founder XT cells (OSE4 and ES-2 cells).
The HEK293FT cells (Invitrogen) were used to produce lentivirus using helper plasmids pSPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 12259). After transfecting with the targeting vectors (pLentiXT1 mCherry or pLentiXT2 mCherry), the virus was collected and used to infect cells plated in six-well plates at six serial dilutions. Cells were re-plated in 10 cm dishes one day after infection and selected with hygromycin B (50–300 µg/mL, depending on cell lines; Mediatech, Manassas, VA) two days after infection. Single clones that were mCherry-negative in the absence of doxycycline (Sigma-Aldrich, St. Louis, MO) and mCherry-positive 24 h after addition of 1 µg/mL doxycycline were expanded using cloning cylinders. An inverted fluorescence microscope (Eclipse TE200, Nikon, Melville, NY) was used to identify the desired clones. The clones produced from cells infected with the lowest amount of virus were selected; these cells generally represented <2% cells infected by virus.
Recombinase-mediated cassette exchange (RMCE)
Founder XT cells were transfected with 1 µg donor vector and 0.1 µg pFIC using the Lipofectamine LTX reagent (Life Technologies) in one well of a six-well plate. Cells were re-plated one or two days after transfection, then were subject to puromycin (Sigma-Aldrich) selection (1–3 µg/mL) four or five days after transfection. Two days after puromycin selection, live cells were pooled for expansion and doxycycline induction.
Flow cytometry and Western blotting
Flow cytometry was performed using a BD LSR II Flow Cytometer (BD Biosciences, San Jose, CA). The antibodies used in Western Blot analysis were: ARID1A (HPA005456, Sigma-Aldrich) and GAPDH (G9545, Sigma-Aldrich).
Results and discussion
We constructed a unique set of plasmids using PCR and cloning to test our methodology for establishing inducible cell lines (). All of the targeting and donor vectors contain the LoxP and FRT sites recognized by Cre and Flp recombinases, respectively, and therefore were named “XT-plasmids”. The targeting vector pLentiXT1 mCherry contains the CMV/TO promoter and the mCherry gene, whose transcription is blocked by binding of TR to the tetracycline operator sequences (TO or TetO2) in the absence of tetracycline. Tetracycline and its derivative doxycycline bind to TR and change the conformation of TR to release from TetO2 sites, thus derepressing transcription from the CMV promoter (T-REx system, Invitrogen, Life Technologies). The mCherry cDNA and the hygromycin resistant gene are flanked by LoxP and FRT sites. This LoxP-FRT cassette is followed by a puromycin resistance gene (Puro) lacking the ATG start codon. The targeting vector pLentiXT2 mCherry differs from pLentiXT1 mCherry in that its Tet-responsive elements are composed of the TRE-minCMV (tetracycline-response element-minimal CMV) promoter and rtTA3 (reverse tetracycline transactivator 3) from the Tet-On system (Clontech) (). In this system, the TRE-minCMV is activated in the presence of both rtTA3 and tetracycline (or doxycycline). The donor vector pXT series can be used for cloning the SOI for overexpression or knockdown, and the resulting pXT-SOI plasmids serve as donors for RMCE ().
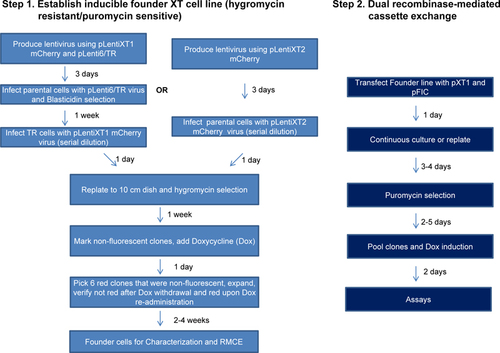
The XT-Cell method consists of two steps (). First, a founder reporter line is established by single-cell cloning after lentiviral transduction of pLenti6/TR (for expressing TR) and the targeting vector. The fluorescent mCherry protein facilitates single-cell cloning for clones that have the best inducibility (i.e., low expression without tetracycline and high expression after addition of tetracycline). The founder cells are resistant to hygromycin but sensitive to puromycin. As the second step, the donor vector was transfected into the founder cells along with a vector (pFIC) expressing Cre and Flp recombinases. Subsequently, puromycin was used to select those cells with successful RMCE reactions.
As proof of principle, we expanded a HeLa-XT1 founder reporter cell line from a single clone after transducing HeLa cells with pLenti6/TR and pLentiXT1 mCherry viruses, with the aid of an inverted fluorescence microscope. A serial dilution of the targeting lentivirus was prepared to infect cells, and the dilution at which <2% of cells were infected was selected for expansion of the founder lines. The clones were selected and expanded based on undetectable mCherry signal in the absence of doxycycline, strong fluorescence upon addition of doxycycline, and undetectable fluorescence after doxycycline withdrawal. Approximately 60% of HeLa cell clones grown in culture dishes appeared to satisfy the criteria above, albeit with variable fluorescence signals upon induction. Flow cytometric analysis confirmed low basal level of mCherry expression in the absence of doxycycline, and >95% of cells exhibited homogenous mCherry fluorescence signal after doxycycline induction in the clone selected (). The donor vector pXT1-GFP was then co-transfected with pFIC vector expressing Cre and Flp recombinases into the HeLa-XT1 mCherry cells. Puromycin was then administrated to select those cells with successful RMCE reactions. As expected, the majority of the puromycin-selected cells retained Tet-inducibility for GFP after RMCE, with homogenous GFP expression observed in >95% of cells upon doxycycline administration (). We noticed that a small fraction of cells (<1%) still expressed mCherry after puromycin selection (), perhaps due to endogenous development of puromycin resistance. We consider that the small percentage of cells would not likely interfere with most biochemical and cell biology assays. Using this method, we were able to establish inducible cell lines (obtaining >1 million cells) including HeLa, HEC-1-A, ES-2, and OSE4, which expressed SOIs in 7–10 days with minimal hands-on time, after establishing the founder XT cells (Supplementary Figure S1).
(A) mCherry expression in HeLa-XT1 mCherry cells were analyzed using flow cytometry, in the absence or presence of doxycycline. (B) GFP expression was analyzed in HeLa-XT1 GFP cells that arise from RMCE. Doxycycline (0.5 µg/mL) was added in culture media for 2 days before flow cytometry analyses. (C) Western blots showing ARID1A expression in the HeLa-XT1 ARID1A cells 2 days after 0.5 µg/mL doxycycline induction. (D) Western blots showing ARID1A expression in the HeLa-XT1 shARID1A cells 2 days after 0.5 µg/mL doxycycline induction. GAPDH serves as a loading control. Note that the ARID1A signal in (D) was overexposed.
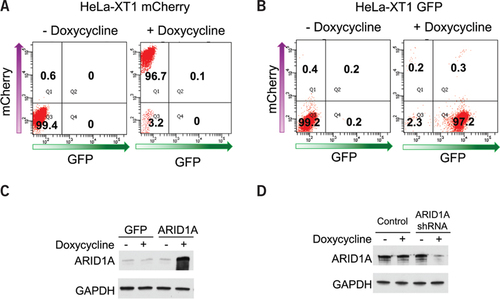
In another example, we used ARID1A as the gene of interest because it is a recently identified tumor suppressor gene whose cDNA is over 6.6 kb (Citation13). The size of the DNA that can be introduced into the XT cells using RMCE is limited by the capacity of the donor vector, and large DNA constructs can be efficiently introduced, as shown by successful ARID1A expression (). The XT founder cells are also efficient for establishing inducible knockdown cell lines when incorporating the microRNA-adapted shRNA. We cloned shRNA targeting ARID1A into plasmid pXT1 shRNA and transfected the plasmid into HeLa-XT1 mCherry cells with pFIC. In the resulting HeLa-XT1 shARID1A cells, ARID1A shRNA was robustly induced and produced a significant decrease in protein expression ().
To demonstrate that successful RMCE reactions can occur in the cells after puromycin selection, we designed primers spanning the LoxP and FRT sites that were then used for PCR with genomic DNA isolated from the HeLa and OSE4 XT cells. The PCR products were observed at expected sizes, suggesting precise recombinations have occurred at both the LoxP and FRT sites in a majority of puromycin-resistant cells after RMCE (Supplementary Figure S2). To estimate the rate of successful RMCE reaction using the XT-cell method, we counted the colony formation units of HeLa-XT1 GFP cells in conditions with various ratios of donor and pFIC vectors. In the absence of pFIC, which expresses both Cre and Flp recombinases, no recombination occurs as evidenced by the lack of puromycin resistant clones (). When a 10:1 ratio of pXT1 GFP:pFIC was used during transfection, the number of clones was highest among the conditions tested, and it was estimated that at least 1.7% of cells underwent successful RMCE ().
Table 1. Optimization of RMCE efficiency by altering the amount of recombinases expressed.
We hypothesized that the genomic locus where the Tet-responsive promoter is inserted determines its inducibility and that Tet-inducibility would be retained after replacing the fluorescence reporter with another SOI. The targeting vectors used to create founder lines were designed such that the Tet-responsive promoter is not flanked by the recombinase-recognition sites and therefore stays in the host genome. This is distinct from previous studies where both the target SOI and the Tet-responsive promoter are provided by the donor vectors and exchanged into the genome (Citation3–5). The fluorescent protein mCherry facilitates the selection of clones where the Tet-responsive promoter was inserted into genome in a way that produces the best inducibility.
Use of a fluorescence protein reporter to screen for ideal founder cells with optimal induction characteristics has been reported by Brough et al. (Citation15). However, instead of employing the cassette exchange strategy shown here, only one recombinase recognition site was introduced in the founder cells to insert a whole plasmid containing a transgene (Citation15). Using the ScIn-2 method, two sequential transfection steps with Cre and Flp recombinases and two single-cell cloning steps were needed to establish Tet-inducible cell lines after identifying an ideal founder cell line (Citation15). In addition, the co-introduction of prokaryotic vector parts may trigger uncontrolled heterochromatization (Citation15, Citation16). Our system avoids these drawbacks, and our results demonstrate that Tet-inducible cell lines can be efficiently established using the XT-cell method.
The fluorescent protein allows selection of the desired clones under a microscope well before they are expanded to sufficient quantity for other analyses such as immunofluorescence staining or Western Blot. Therefore, the time and effort it takes to establish inducible cell lines using this 2-step XT-cell method can be less than that using the conventional method (). The mCherry vector also allows establishment of the founder cells using fluorescence activated cell sorting. However, the major time consuming step for this technology is establishing the founder lines (). Once the founder lines are established, many inducible cell lines for any SOI can be created quickly using our system. All of the different cell lines after RMCE are derived from a particular founder cell line. This means they are “isogenic” in the sense that the transgenes all have the same Tet-inducible promoter at the same genomic insertion site. Therefore, this method would be particularly useful when comparing the phenotypes of multiple genes or multiple mutations in a given gene.
The current procedures for establishing founder XT cells are time-consuming and labor intensive. However, after creating founder lines through collaborative efforts, these cell lines can be used to establish doxycycline-inducible cells containing the sequence of interest quickly with minimal hands-on time.
One limitation of the current study is that we did not perform Southern blot analysis to unequivocally demonstrate that the founder cell lines contain only one copy of the targeting vectors. Instead, we took advantage of the proven property of single-integration of lentivirus when used at a low multiplicity of infection (Citation16) to obtain XT founder reporter cells. Under this condition, the founder cells presumably contain only a single integration of the targeting vectors. Our success with the RMCE procedure also suggests that the founder cells tested only underwent one integration event (Supplementary Figures S1 and S2). Therefore, Southern blotting could be omitted in the shortened protocol for the XT-cell method, although it would be helpful to confirm that the founder cells only harbor one copy of the targeting vectors. The second limitation of our method is that the founder cell lines may differ, genetically and epigenetically, from their parental cells due to single-cell selection and expansion. Careful characterization of the founder cell lines could identify the ideal founder cell lines that not only have optimum induction characteristics and RMCE efficiency, but also are as close as possible to the parental cells genetically and phenotypically. Establishing such founder cells would require significant effort. Therefore, we propose a process of setting a collaborative initiative to create a collection of founder XT reporter lines, by creating an online portal that lists the available XT cell lines. All cell lines shall be quality checked by short-tandem repeat profiling, genomic integration position identification, similar proliferation rates/morphology, and similar genomic profiles compared with parental lines. The founder cell lines and the XT plasmid set will be available for academic research laboratories. We believe that our strategy, by facile modulation of gene expression in cell culture systems, could expedite many aspects of biomedical research in this field.
Author contributions
BG designed and performed experiments, analyzed data, and wrote the manuscript. TM performed cloning for some of the XT plasmids. TLW and IMS wrote the manuscript.
Competing interests
BG, IMS and TLW filed an invention disclosure for the XT-cell method.
Supplementary Material For: Establishing isogenic inducible cell lines using founder reporter lines and recombinase-mediated cassette exchange
Download PDF (537.8 KB)Acknowledgments
We thank Yuri Voziyanov for the generous gift of the pFIC plasmid. This work was supported by the National Institutes of Health [R21CA165807, RO1CA103937, RO1CA129080, and RO1CA148826], and the Ann Schreiber Research Training Programs of Excellence grant [POE/JHU/01.12] awarded to B.G. by the Ovarian Cancer Research Fund and the Tell Every Amazing Lady Louisa M. McGregor Ovarian Cancer Foundation. This paper is subject to the NIH Public Access Policy.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2144/000114098
Additional information
Funding
References
- Markstein, M., C.Pitsouli, C.Villalta, S.E.Celniker, and N.Perrimon. 2008. Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet.40:476–483.
- Meerbrey, K.L., G.Hu, J.D.Kessler, K.Roarty, M.Z.Li, J.E.Fang, J.I.Herschkowitz, A.E.Burrows, et al.. 2011. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. USA108:3665–3670.
- Weidenfeld, I., M.Gossen, R.Low, D.Kentner, S.Berger, D.Gorlich, D.Bartsch, H.Bujard, and K.Schonig. 2009. Inducible expression of coding and inhibitory RNAs from retargetable genomic loci. Nucleic Acids Res.37:e50.
- Wong, E.T., J.L.Kolman, Y.C.Li, L.D.Mesner, W.Hillen, C.Berens, and G.M.Wahl. 2005. Reproducible doxycycline-inducible transgene expression at specific loci generated by Cre-recombinase mediated cassette exchange. Nucleic Acids Res.33:e147.
- Khandelia, P., K.Yap, and E.V.Makeyev. 2011. Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc. Natl. Acad. Sci. USA108:12799–12804.
- Schlake, T. and J.Bode. 1994. Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci. Biochemistry33:12746–12751.
- Cheon, D.J. and S.Orsulic. 2011. Mouse models of cancer. Annu. Rev. Pathol.6:95–119.
- McMullin, R.P., L.N.Mutton, and C.J.Bieberich. 2009. Hoxb13 regulatory elements mediate transgene expression during prostate organogenesis and carcinogenesis. Dev. Dyn.238:664–672.
- Soyal, S.M., A.Mukherjee, K.Y.S.Lee, J.Li, H.G.Li, F.J.DeMayo, and J.P.Lydon. 2005. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis41:58–66.
- Campeau, E., V.E.Ruhl, F.Rodier, C.L.Smith, B.L.Rahmberg, J.O.Fuss, J.Campisi, P.Yaswen, et al.. 2009. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE4:e6529.
- Nam, H.S. and R.Benezra. 2009. High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem Cell5:515–526.
- Anderson, R.P., E.Voziyanova, and Y.Voziyanov. 2012. Flp and Cre expressed from Flp-2A-Cre and Flp-IRES-Cre transcription units mediate the highest level of dual recombinase-mediated cassette exchange. Nucleic Acids Res.40:e62.
- Guan, B., T.L.Wang, and M.Shih Ie. 2011. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res.71:6718–6727.
- Barry, S.C., B.Harder, M.Brzezinski, L.Y.Flint, J.Seppen, and W.R.Osborne. 2001. Lentivirus vectors encoding both central polypurine tract and posttranscriptional regulatory element provide enhanced transduction and transgene expression. Hum. Gene Ther.12:1103–1108.
- Brough, R., A.M.Papanastasiou, and A.C.Porter. 2007. Stringent and reproducible tetracycline-regulated transgene expression by site-specific insertion at chromosomal loci with pre-characterised induction characteristics. BMC Mol. Biol.8:30.
- Turan, S., C.Zehe, J.Kuehle, J.Qiao, and J.Bode. 2013. Recombinase-mediated cassette exchange (RMCE) - a rapidly-expanding toolbox for targeted genomic modifications. Gene515:1–27.

