
Abstract
Here we describe a method for high-throughput genotyping of live larval zebrafish as early as 72 h post-fertilization (hpf). Importantly, this technique allows rapid and cost-effective PCR-based genotyping from very small fin biopsies, which regenerate as the embryo develops, thereby allowing researchers to select embryos with desired genotypes to be raised to adulthood.
Keywords: :
This method enables accurate genotyping of zebrafish embryos as early as three days post-fertilization via PCR-based analysis of genomic DNA extracted from embryonic fin clips using alkaline lysis. The fin clip procedure is compatible with normal embryonic development and reduces the number of fish that must be raised to adulthood.
The advent of genome editing technologies such as TALENs (Citation1–3), and more recently the CRISPR-CAS9 system (Citation4), has led to a global acceleration in the production of targeted knockouts among the zebrafish community. However, a persistent bottleneck in generating zebrafish knockouts is the time required to identify transmitting founder fish and subsequent carriers via traditional fin clip approaches involving adult zebrafish. Here we present an embryonic fin clip strategy, which will expedite the latter step.
Previous strategies for genotyping live zebrafish embryos have employed proteinase K digestion, which leads to variable PCR efficiency and permits only a small number of alleles or transgenes to be genotyped per embryo (Citation5). Our approach enables the genotyping of live zebrafish embryos as early as 72 hpf via transection of the embryonic tail fin, followed by rapid genomic DNA extraction and genotyping by PCR and/or restriction enzyme digestion. Importantly, this embryonic fin clip strategy has a high PCR efficiency, a high survival rate, and a low rate of false positives. Furthermore the high-throughput nature of this protocol enables genotyping of large numbers of embryos before they reach free feeding stage at 5.2 days post-fertilization (dpf), when they become protected by government legislation in many countries. We find it is routinely possible to fin clip 96 embryos in 2.5 h using this approach.
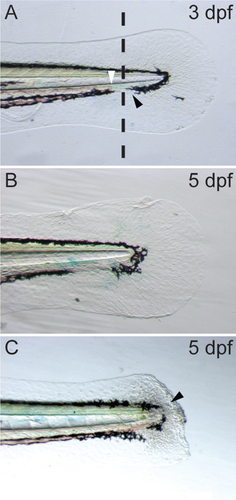
Embryos are anesthetized in Tricaine as described previously (Citation6), and 3 µL 20% TWEEN 20 is added to each Petri dish (30 mL) to aid in transfer of resultant fish biopsies. Embryos are transferred using a P20 micropipettor and a cut-off tip onto the lid of a Petri dish that has been lined with a strip of autoclave tape. The lid is then transferred to a stereomicroscope under top illumination and excess surrounding liquid is pipetted off. The tip of the caudal fin is removed using a microscalpel (#500249; World Precision Instruments, Sarasota, FL) by applying steady downward pressure to make the incision (, dotted line) within the pigment gap (, black arrowhead) distal to the circulating blood (, white arrowhead). This prevents bleeding and maximizes embryo survival. We consistently achieve 100% survival of embryos following this procedure from the time of fin clip at 3 dpf, to the time they enter the aquarium to be raised at 5.2 dpf (n = 571 embryos, 9 independent procedures). The fin clip procedure induces blastema formation at the site of biopsy (, black arrowhead), following which the caudal fin regenerates and normal development proceeds. The fin tissue remains embedded in the autoclave tape, which allows the biopsied embryo to be transferred to a 24 well plate, each well containing 1 mL E3 medium where the embryo may be incubated for several days while genotyping is carried out.
(A) Dotted line represents the site of transection at three days post-fertilization (dpf), black arrowhead highlights the pigment gap; white arrowhead, highlights the caudal limit of blood circulation. (B) Untransected embryonic fin at five dpf. (C) Embryonic zebrafish fin at five dpf, two days post-transection. Black arrowhead highlights blastema formation and ongoing fin regeneration.
Using a protocol modified from Meeker et al. (Citation7), the fin biopsy is then transferred to a 96 well plate containing 15 µL 50 mM NaOH using a P20 pipettor with a standard tip. The sample is heated at 98°C for 2 min, cooled down and neutralized with 1/10th volume of 1M TRIS-HCl, pH8.0, followed by brief centrifugation to pellet cellular debris. To demonstrate the efficiency of the fin clip protocol, we genotyped the progeny of a vhlhu2117 heterozygous incross (Citation8) (Primers 5′-TAAGGGCTTAGCGCATGTTC-3′; 5′-CGAGTTAAACGCGTAGATAG-3′). The vhlhu2117 mutation destroys a BciVI site, which permits genotyping via restriction fragment length polymorphism. Indeed many genome-editing technologies routinely employ this approach to aid in the identification of adult founders and carriers. We have used this technique to genotype embryos from many different genetic backgrounds; the PCR efficiency from 9 independent experiments is 97% (n = 555/571).
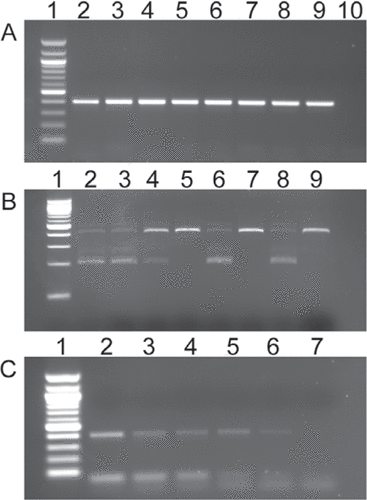
The genotyping PCR generates a 412 bp fragment (), which when digested, releases fragments of approximately 200 bp when the BciVI restriction site is intact in the wild-type allele (). The presence of a single 200 bp band indicates a wild-type embryo, while a 412 bp and a 200 bp band indicate heterozygosity of the vhlhu2117 allele (). It follows that the presence of a single undigested 412 bp band indicates a vhl mutant. Eight randomly selected embryos were biopsied following the protocol above and 1 µL of the resulting lysate was used as template. The PCR conditions were as follows, 94°C 4 min, 92°C 30 s, 56°C 30 s, 72°C 40 s, 40 cycles, 72°C 5 min. From a 20 µL reaction volume, 5 µl was analyzed on a 1% agarose/TAE gel (); lanes 2–9 represent a single fin clip lysate in each case, and lane 10 is a DNA negative control. A 100 bp ladder (NEB) is shown in lane 1. To digest the PCR reaction, 5 µL was mixed with 2 µL NEB buffer 4, 0.5 µL BciVI (10 U/ µl and made up to 20 µL with distilled water. This mixture was incubated at 37°C for 3 h in a PCR block.
In each case, lane 1 contains a 100 bp DNA ladder (NEB). (A) PCR amplification of vhl mutant region results in a 412 bp DNA fragment. Lanes 2–9 contain PCR products generated from lysates from individual embryonic fin biopsies. Lane 10 contains a DNA negative control. (B) Digestion of the PCR products shown in panel A with BciVI identifies vhl heterozygous embryos (lanes 2, 3, 4, 6, 8) and vhl mutants (lanes 5, 7, 9). (C) Dilution series of the embryonic fin biopsy lysate used in panel A. Lanes 2–6 depict 1:2, 1:10, 1:20, 1:50, and 1:100 dilutions respectively. Lane 7 contains a DNA negative control.
Samples were analyzed on a 3% high-resolution gel containing 1.5% MetaPhor agarose (Lonza, Basel, Switzerland) and 1.5% standard agarose (). The presence of both 200 bp and 412 bp bands in lanes 2, 3, 4, 6, and 8 indicates vhl heterozygous embryos, while the absence of 200 bp bands in lanes 5, 7, and 9 indicates vhl mutant embryos (). Vascular looping abnormalities characteristic of vhl mutant embryos were present in embryos corresponding to lanes 5, 7, and 9 at 5 dpf and absent from all other embryos (Citation9). It is worth noting that the presence of bands running higher than 412 bp in lanes 2, 3, 4, 6, and 8 indicate DNA heteroduplex formation between mutant and wild type strands. These are notably absent from lanes 5, 7, and 9. It is also important to note that in , the 412 bp band appears more intense, while in other lanes the 200 bp band appears more intense (compare lane 4, with lanes 2, 3, 6, 8). This could suggest cross contamination; however, we routinely use this approach to identify homozygous embryos for raising. From 3 independent experiments utilizing 3 distinct homozygous viable lines, we have successfully raised 25/30 (83%) homozygous embryos to adulthood (>6 months). Of the 25 surviving embryos, 24/25 (96%) were confirmed as homozygotes either by adult fin clip or by genotyping of maternal zygotic embryos. A single false positive fish was identified as a heterozygous carrier. This indicates that any cross contamination has a minimal effect on genotyping efficiency, although care must be taken if using this protocol to identify transgenic embryos, where the presence or absence of a PCR product is used as a means of identification.
To give an approximation of the theoretical total number of alleles that can be genotyped from a single embryonic fin biopsy, a dilution series was made of the embryo lysate used in the PCR shown in lane 2 (). Dilutions of 1:2, 1:10, 1:20, 1:50, and 1:100 were set up, and 1 µL of each dilution was used as a template to amplify the vhl allele using the above PCR conditions, in this case for only 35 cycles. Lane 1 contains a 100 bp DNA ladder, while lanes 2–6 contain the dilutions in order of decreasing concentration. Lane 7 contains a DNA negative control. Again, 5 µL of a 20 µL reaction was analyzed on a 1% agarose/ TAE gel. Clearly, even a 100-fold dilution of the sample produced an amplification product. Given that the total volume of the embryonic lysate was approximately 18 µL (15 µL 50mM NaOH, 1.5 µL transfer volume, 1.5 µL 1M Tris pH 8.0), this gives a theoretical value of 1800 alleles that could be amplified from a single embryonic fin biopsy. Although this number will vary depending upon the efficiency of a particular reaction, the result demonstrates the robustness of the technique for generating high quality PCR-ready genomic DNA.
Author contributions
RNW designed and performed research, analyzed the data, and also wrote and edited the paper. SE designed and performed research. FJMvE and PWI edited the paper.
Competing interests
The authors declare no competing interests.
Fin clipping and genotyping embryonic zebrafish at 3 days post-fertilization
Download PDF (162.1 KB)Acknowledgments
This research was supported by a ZF-Health award (EC-FP7 HEALTH-F4-2010-242048 F.J.M.v.E), the Royal Society (Research grant RG120564 R.N.W), The Wellcome Trust (program grant 082962/Z/07/Z; P.W.I. and F.J.M.v.E.), and by the JG Graves Medical Research Fellowship awarded to R.N.W.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2144/000114116
Additional information
Funding
References
- Bedell, V.M., Y.Wang, J.M.Campbell, T.L.Poshusta, C.G.Starker, R.G.Krug, 2nd, W.Tan, S.G.Penheiter, et al.. 2012. In vivo genome editing using a high-efficiency TALEN system. Nature491:114–118.
- Sander, J.D., L.Cade, C.Khayter, D.Reyon, R.T.Peterson, J.K.Joung, and J.R.Yeh. 2011. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol.29:697–698.
- Zu, Y., X.Tong, Z.Wang, D.Liu, R.Pan, Z.Li, Y.Hu, Z.Luo, et al.. 2013. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat. Methods10:329–331.
- Hwang, W.Y., Y.Fu, D.Reyon, M.L.Maeder, S.Q.Tsai, J.D.Sander, R.T.Peterson, J.R.Yeh, and J.K.Joung. 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol.31:227–229.
- Kawakami, K. and N.Hopkins. 1996. Rapid identification of transgenic zebrafish. Trends Genet.12:9–10.
- Westerfield, M. 1993. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). Univ. of Oregon Press, Eugene.
- Meeker, N.D., S.A.Hutchinson, L.Ho, and N.S.Trede. 2007. Method for isolation of PCR-ready genomic DNA from zebrafish tissues. Biotechniques43:610–614.
- van Rooijen, E., E.E.Voest, I.Logister, J.Korving, T.Schwerte, S.Schulte- Merker, R.H.Giles, and F.J.van Eeden. 2009. Zebrafish mutants in the von Hippel- Lindau tumor suppressor display a hypoxic response and recapitulate key aspects of Chuvash polycythemia. Blood113:6449–6460.
- van Rooijen, E., E.E.Voest, I.Logister, J.Bussmann, J.Korving, F.J.van Eeden, R.H.Giles, and S.Schulte-Merker. 2010. von Hippel-Lindau tumor suppressor mutants faithfully model pathological hypoxia-driven angiogenesis and vascular retinopathies in zebrafish. Disease models & mechanisms3:343–353.

