
Abstract
The complete genome of the bacterial pathogen Pseudomonas aeruginosa has now been sequenced, allowing gene deletion, one of the most frequently used methods in gene function study, to be fully exploited. In this study, we combine the sacB-based negative selection system with a cre-lox antibiotic marker recycling method. This methodology allows allelic exchange between a target gene and a gentamicin cassette flanked by the two lox sequences. A tetracycline plasmid expressing the cre recombinase is then introduced in the mutant strain to catalyze the excision of the lox-flanked resistance marker. We demonstrate here the efficiency of the combination of these two methods in P. aeruginosa by successively deleting ExoS and ExoT, which are two genetically independent toxins of the type-three secretion system (TTSS). This functional cre-lox recycling antibiotic marker system can create P. aeruginosa strains with multiple mutations without modifying the antibiotic resistance profile when compared to the parental strain.
Introduction
Since the complete genome of Pseudomonas aeruginosa has been sequenced (Citation1) substantial progress has been made in the study of this Gram-negative opportunistic pathogen, which is associated with chronic lung injury in cystic fibrosis (CF) patients and with septicemia in burned or neutropenic patients (Citation2–4). Study of gene function is often carried out by deleting genes in the strains of interest and observing the phenotypic differences between the parental and deleted strains. Antibiotic markers that allow the selection of the recombinant clone usually replace these genes (Citation5). This allelic exchange method has two major disadvantages when working with P. aeruginosa. First, since P. aeruginosa rapidly develops spontaneous antibiotic resistance, there are no more than two or three antibiotic resistance cassettes available for allelic exchanges. Second, generating double-mutant strains leads to manipulated strains with a dangerous antibiotic resistance background. Marx and Lidstrom (Citation6) recently described a method for marker recycling in Gram-negative bacteria, namely Methylobacterium extorquens and Burkholderia fungorum, based on the cre-lox methodology. They show that lox-flanked antibiotic markers can be removed from the bacterial genome after the allelic exchange. The site-specific recombinase encoded by the cre gene from the P1 phage catalyzes the in vivo excision of DNA sequences flanked by two lox recognition sites (Citation7–9). In this study, we have combined the sacB-based negative selection methodology described by Schweizer and Hoang (Citation5) with the cre-lox system of Marx and Lidstrom (Citation6), in order to generate a multiple mutant strain of P. aeruginosa with no resistance marker. To validate this method for marker recycling, we deleted the two major toxins secreted by the type III secretion system (TTSS) of P. aeruginosa, ExoS and ExoT, in the already described CHA parental strain (CF isolate) (Citation10).
Materials and methods
Bacterial Strains and Growth Conditions
Bacterial strains and plasmids used in this study are listed in . All bacteria were grown from single-colony isolates or overnight cultures in Luria-Bertani (LB) broth with 10 g/L NaCl. Liquid cultures were grown at 37°C under air with shaking at 300 rpm. Media were solidified with 1.2% Bacto Agar (Difco, Pont de Claix, France). Bacterial growth was monitored spectrophotometrically at an absorbance of 600 nm (A600). Antibiotics were used for Escherichia coli at the given concentrations: 100 µg/mL ampicillin, 10 µg/mL gentamicin, and 10 µg/mL tetracycline. P. aeruginosa strains were plated on Pseudomonas isolation agar (Difco) supplemented with 600 µg/mL carbenicillin, 250 µg/mL tetracycline, or 400 µg/mL gentamicin. Liquid cultures of P. aeruginosa were supplemented with 300 µg/mL carbenicillin, 200 µg/mL gentamicin, or 250 µg/mL tetracycline. P. aeruginosa type III secretion was analyzed after dilution to an A600 of 0.2 of a culture grown overnight (at 37°C with aeration) supplemented with antibiotics as required. Secretion was then induced upon calcium depletion of the culture with 5 mM EGTA in the presence of 20 mM MgCl2.
Table 1. Bacterial Strains and Plasmids Used in this Study
Vector Construction
We first improved the pEX100T vector (Citation5) by inserting the pUC18 polylinker, generating pEX100Tlink (). To that purpose, the aacC1 gene (gentamycin acetyltransferase-3-1, the gene conferring gentamycin resistance) from pUCGm (Citation11) was cloned in the SmaI site of pUC18, creating the pUC18Gm vector. The aacC1 gene was then extracted from this vector by PvuII digestion and cloned into pEX100T linearized with SmaI. The SacI restriction enzyme was then used to eliminate aacC1 in order to leave only one multicloning site (MCS) in pEX100T.
(A) Plasmid map of the allelic exchange vector pEX100Tlink. pEX100Tlink is derived from the pEX100T suicide vector (GenBank accession no. U17500) with the addition of a multicloning site (MCS). Antibiotic resistance is encoded by bla (ampicillin). The sacB gene confers sucrose sensitivity on the Pseudomonas aeruginosa strain when grown on LB medium supplemented with 5% sucrose. oriT is the origin of transfer for conjugation-mediating transfer, and ori is the pMB1-based origin of replication (Citation5). (B) Plasmid map of the pUCGmlox vector. pUCGmlox is derived from pUCGm (GenBank accession no. U04610). Antibiotic resistances are encoded by bla (ampicillin) and by aacC1 (gentamicin). The aacC1 gene, flanked by the lox sequences, was amplified from pCM351 (GenBank accession no. AY093430). The entire aacC1lox sequence is flanked by a palindromic MCS.
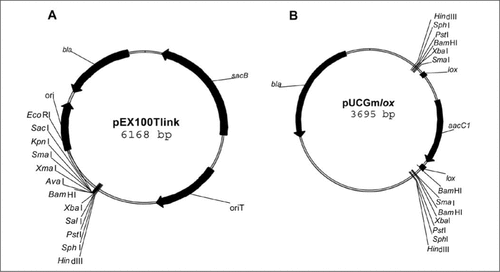
The pUCGmlox () vector was then created as follows. The aacC1 gene flanked with the two lox sequences (GenBank® accession no. E12288) was amplified by PCR from pCM351 (Citation6) using primers containing SacI restriction sites (sequences given in ). pUCGm (Citation11) was then digested with SacI, and the original aacC1 gene was replaced by the lox-flanked aacC1 PCR product.
Table 2. PCR Primers
P. aeruginosa Adaptation of the cre-lox System
The regions immediately flanking exoS and exoT were amplified by PCR using the primers listed in and the high-fidelity polymerase PfuTurbo (Stratagene, Amsterdam, The Netherlands). PCR products exoS- or exoT-upstream and exoS- or exoT-downstream were digested by EcoRI and HindIII or BamHI and HindIII and cloned by a three-way ligation into pEX100Tlink () deleted for the HindIII restriction site and opened by EcoRI and BamHI (). These cloning strategies generated pLQ34 and pLQ35, respectively. The 1 kb aacC1lox gene, extracted from pUCGmlox (), was then cloned in the single HindIII site formed by the ligation between the two flanking fragments, producing plasmids pLQ36 and pLQ37. These donor constructs, containing both the sacB counter-selective marker and the Gmlox antibiotic resistance cassettes, were then transformed into the E. coli S17.1 helper strain (Citation12).
The CHAΔSGmlox mutant and CHAΔTGmlox mutant of P. aeruginosa were generated by respectively introducing pLQ36 and pLQ37 from E. coli S17.1 by conjugation. Allelic exchange of exoS and exoT was conducted as described previously (Citation5). Gentamicin-resistant (GmR) transconjugants obtained were counter-selected on LB medium containing 5% sucrose. Sucrose-resistant colonies were screened for GmR and carbenicillin sensitivity (CbS) to identify double recombinants. One such mutant was chosen for further characterization.
The pCM157 plasmid (Citation6) was then introduced by electroporation as described previously (Citation13) into these two mutant strains. Its IncP origin of replication (oriV) allows it to be maintained in P. aeruginosa. One tetracycline-resistant (TcR) transconjugant clone from each mutant was grown overnight in LB supplemented with tetracycline in order to allow the expression of the cre recombinase. The pCM157 was then cured from the strain by three successive growth cycles in LB broth without tetracycline. The selected clone for each mutant, CHAΔSlox and CHAΔTlox, were sensitive for gentamicin and tetracycline.
A strain deleted for exoS and exoT was generated by transforming pLQ37 into the unmarked CHAΔSlox background. The double-mutant strain obtained, CHAΔSTGmlox, possessed the aacC1lox resistance marker only in the exoT allele. As described above, Cre recombinase was used to recycle the resistance cassette.
Analysis of Secreted Fractions
Overnight, cultures of wild-type or mutant strains were diluted (A600=0.2) in 4 mL LB broth either in induced or control (non-induced) conditions. Bacteria were grown at 37°C under air with shaking at 300 rpm for an additional 3 h. Supernatant fractions normalized to an A600 of 1.8 were prepared by pelleting 1.5 mL culture for 5 min at 12,000× g. Supernatant fractions (1 mL) were then transferred to a fresh microcentrifuge tube, and proteins were precipitated by adding 350 µL 50% trichloroacetic acid. After a 30-min incubation on ice, precipitated proteins were collected by centrifugation (10 min at 12,000× g), washed with 1 mL ice-cold acetone, and suspended in 20 µL sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. Supernatants were separated on a 10% SDS-PAGE gel, and proteins were stained with Coomassie® blue.
Results and discussion
shows the two plasmids used in the strategy: (i) pEX100Tlink, an allelic exchange vector allowing sacB-based negative selection and (ii) pUCGmlox, a convenient source of lox-flanked aacC1. Flanking sequences upstream and downstream of the gene to be deleted are amplified and inserted by three-way ligation into pEX100Tlink. Lox-flanked aacC1 is then inserted at a unique site between these two inserts. Upon transformation and allelic exchange, and given that the allelic exchange vector cannot replicate, positive colonies should be GmR, sucrose-resistant, and CbS. After introduction of Cre recombinase, the GmR marker is removed.
In order to validate this strategy, two unmarked mutants and a double unmarked mutant were generated in the CHA P. aeruginosa strain. Allelic exchange and recycling of the resistance marker (Figure 2A) were monitored by PCR. As shown in , PCR amplification of the intact exoS or exoT allele generated products of 2.4 and 3.4 kb, respectively, for the CHA wild-type strain. Deletion of the gene of interest and insertion of the resistance marker led to a small decrease in the length of the amplified fragment. Recycling the resistance marker and removing 1 kb from the genomic DNA of the strains led to amplification of a 1.1-kb fragment for the deleted exoS locus and a 2.3-kb fragment for the deleted exoT locus.
(A) Strategy for allelic exchange and antibiotic marker recycling. Allelic exchange leads to an aacC1lox deletion/insertion (1 kb) mutant that can then be unmarked through the introduction of the cre expression plasmid pCM157. The process can then be repeated with a second target gene to generate double mutations. Sizes correspond to PCR amplification between primers (black arrows). (B) PCR analysis of allelic exchange and subsequent marker removal for exoS and exoT loci. The forward primer for amplification of the upstream flank and the reverse primer for the downstream flank were used together to amplify across the entire locus. The molecular weight standard in the first lane is a 1-kb DNA Ladder (Promega France, Charbonnieres, France). (C) Secretion profiles of wild-type (WT) and mutant strains. Coomassie blue stained 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of supernatant fraction in type III secretion system (TTSS) induced condition (+) or non-induced conditions (−). Type III-related exoproducts and molecular weight standard (LMW-SDS Marker kit; Amersham Biosciences Europe GmbH, Orsay, France) are indicated on the left side of the gel.
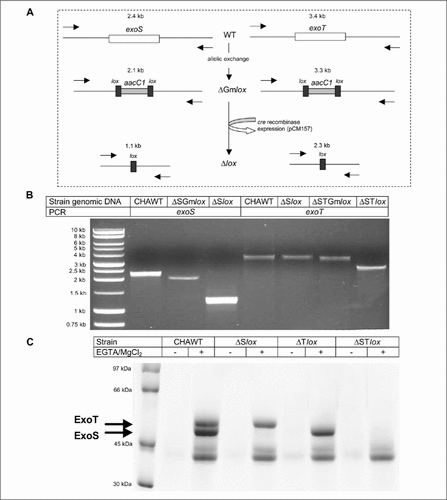
The phenotypic effect of deletion of one or both genes was confirmed by examining the secretion profiles of the three mutants to the wild-type CHA strain (). CHAΔSlox, deleted for the exoS toxin gene, was no longer able to produce and secrete this type III toxin when compared to the wild-type strain that exhibited a secretion of both toxins ExoT and ExoS at 53 and 49 kDa, respectively (Citation14). CHAΔTlox, deleted for the exoT toxin gene, was no longer able to produce and secrete this type III toxin, and the double-mutant strain CHAΔSTlox lost the ability to secrete both toxins, when compared to the CHA wild-type.
Combination of efficient negative selection against single-crossover exconjugants due to the presence of sacB with the antibiotic marker recycling cre-lox strategy eases creation of mutations in the genomic DNA of P. aeruginosa. Although previous methods have described how to generate gene-deleted mutants of Gram-negative bacteria without adding antibiotic markers (Citation6), the negative selection step increases the recovery rate of desired recombinants and thus makes the process more convenient. In addition, this system allows construction of bacterial strains harboring multiple gene deletions.
When performing iterative gene deletion, one must be careful since recombination between lox sequences in the genome can lead to the deletion of large domains (Citation15) instead of the planned one. Nevertheless, for distant deletion loci, we assume that such an event could directly lead to cell death because of the loss of vital genetic information. In case of close loci, deleted sequences have to be exactly characterized.
We assume that this combined method for allelic exchange and antibiotic marker recycling can be used in all the Gram-negative bacterial species in which the sacB system is functional, pEX100Tlink is not able to replicate, and pCM157 can allow the expression of the cre recombinase at a sufficient level for lox recombination.
Competing Interests Statement
The authors declare no competing interests.
Acknowledgments
We would like to thank Prof. Schweizer for providing the pEX100T and pUCGm vectors. We are grateful to Prof. Lidstrom for providing the pCM157 and pCM351 vectors. This work and L. Quénée were supported by the association “Vaincre la Mucoviscidose.” We also thank L. Northrup for linguistic corrections of the manuscript.
Additional information
Funding
References
- Stover, C.K., X.Q.Pham, A.L.Erwin, S.D.Mizoguchi, P.Warrener, M.J.Hickey, F.S.L.Brinkman, W.O.Hufnagle, et al.. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature406:959–964.
- Banwart, B., M.L.Splaingard, P.M.Farrell, M.J.Rock, P.L.Havens, J.Moss, M.E.Ehrmantraut, D.W.Frank, et al.. 2002. Children with cystic fibrosis produce an immune response against exoenzyme S, a type III cytotoxin of Pseudomonas aeruginosa. J. Infect. Dis.185:269–270.
- Moss, J., M.E.Ehrmantraut, B.D.Banwart, D.W.Frank, and J.T.Barbieri. 2001. Sera from adult patients with cystic fibrosis contain antibodies to Pseudomonas aeruginosa type III apparatus. Infect. Immun.69:1185–1188.
- Roy-Burman, A., R.H.Savel, S.Racine, B.L.Swanson, N.S.Revadigar, J.Fujimoto, T.Sawa, D.W.Frank, et al.. 2001. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J. Infect. Dis.183:1767–1774.
- Schweizer, H.P. and T.T.Hoang. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene158:15–22.
- Marx, C.J. and M.E.Lidstrom. 2002. Broad-host-range cre-lox system for antibiotic marker recycling in Gram-negative bacteria. BioTechniques33:1062–1067.
- Palmeros, B., J.Wild, W.Szybalski, S.Le Borgne, G.Hernandez-Chavez, G.Gosset, F.Valle, and F.Bolivar. 2000. A family of removable cassettes designed to obtain antibiotic-resistance-free genomic modifications of Escherichia coli and other bacteria. Gene247:255–264.
- Ayres, E.K., V.J.Thomson, G.Merino, D.Balderes, and D.H.Figurski. 1993. Precise deletions in large bacterial genomes by vector-mediated excision (VEX). The trfA gene of promiscuous plasmid RK2 is essential for replication in several Gram-negative hosts. J. Mol. Biol.230:174–185.
- Sternberg, N. and D.Hamilton. 1981. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol.150:467–486.
- Toussaint, B., I.Delic-Attree, and P.M.Vignais. 1993. Pseudomonas aeruginosa contains an IHF-like protein that binds to the algD promoter. Biochem. Biophys. Res. Commun.196:416–421.
- Schweizer, H.D. 1993. Small broad-host-range gentamycin resistance gene cassettes for site-specific insertion and deletion mutagenesis. BioTechniques15:831–834.
- Simon, R., U.Priefer, and A.Puhler. 1983. A broad rang mobilization system for in vivo genetic ingineering: transposon mutagenesis in Gram-negative bacteria. BioTechnology1:784–791.
- Enderle, P.J. and M.A.Farwell. 1998. Electroporation of freshly plated Escherichia coli and Pseudomonas aeruginosa cells. BioTechniques25:954–956.
- Frank, D.W. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol.26:621–629.
- Wilson, J.W., D.H.Figurski, and C.A.Nickerson. 2004. VEX-capture: a new technique that allows in vivo excision, cloning, and broad-host-range transfer of large bacterial genomic DNA segments. J. Microbiol. Methods57:297–308.

