Abstract
Gaucher disease is a rare inborn error of glycosphingolipid metabolism due to deficiency of lysosomal acid β-glucocerebrosidase; the condition has totemic significance for the development of orphan drugs. A designer therapy, which harnesses the mannose receptor to complement the functional defect in macrophages, ameliorates the principal clinical manifestations in hematopoietic bone marrow and viscera. While several aspects of Gaucher disease (particularly those affecting the skeleton and brain) are refractory to treatment, enzyme (replacement) therapy has become a pharmaceutical blockbuster. Human β-glucocerebrosidase was originally obtained from placenta and the Genzyme Corporation (Allston, MA) subsequently developed a recombinant product. After purification, the enzyme is modified to reveal terminal mannose residues which facilitate selective uptake of the protein, imiglucerase (Cerezyme®), in macrophage-rich tissues. The unprecedented success of Cerezyme has attracted fierce competition: two biosimilar agents, velaglucerase-alfa, VPRIV® (Shire Human Genetic Therapies, Dublin, Ireland) and taliglucerase-alfa (Protalix, Carmiel, Israel), are now approved or in late-phase clinical development as potential ‘niche busters’. Oral treatments have advantages over biological agents for disorders requiring lifelong therapy and additional stratagems which utilize small, orally active molecules have been introduced; these include two chemically distinct compounds which inhibit uridine diphosphate glucose: N-acylsphingosine glucosyltransferase, the first step in the biosynthesis of glucosylceramide – a key molecular target in Gaucher disease and other glycosphingolipidoses. Academic and commercial enterprises in biotechnology have combined strategically to expand the therapeutic repertoire in Gaucher disease. The innovative potential of orphan drug legislation has been realized – with prodigious rewards for companies embracing its humanitarian precepts. In the era before enzyme therapy, bone marrow transplantation was shown to correct systemic disease in Gaucher patients by supplying a source of competent donor macrophages. As a radical advance on cell- or protein-replacement techniques, contemporary methods for transferring genes to autologous hematopoietic stem cells, and to the brain, merit further exploration. At present, the inflated pharmaceutical niche of Gaucher disease appears to be resilient, but if the remaining unmet needs of patients are to be convincingly addressed and commercial development sustained, courageous scientific investment and clinical experimentation will be needed.
Clinical profile
Gaucher disease was the first lysosomal disease for which a specific therapy was introduced in the US orphan legislative milieu. The success of Ceredase® and Cerezyme® (Genezyme Corporation, Allston, MA) has driven pharmaceutical investment in other lysosomal diseases which have disabling effects on life quality and survival. A contemporary Western dilemma rests between the provision of high-cost medicines requiring intensive investment for very few patients and the corporate investment required to improve the health of the population by a more radical attack on the causes of common illness.Citation1
Gaucher disease
Definition
Gaucher disease is caused by a functional deficiency of the acid hydrolase, β-glucocerebrosidase, or glucosylceramidase (E.C.3.2.1.45).Citation2,Citation3 The immediate substrates for this enzyme are glucosylceramide and its nonacylated analog, glucosylsphingosine. These glycosphingolipids arise from the digestion of more complex glucosides and gangliosides present in cell membranes. Rare variant forms of Gaucher disease result from deficiency of the sphingolipid activator protein, saposin C.Citation3 Latterly, inherited defects in a recently discovered molecule, LIMP-2, have been associated with an usual phenotype with β-glucocerebrosidase deficiency in the kidney, brain, and other tissues, but not white cells or macrophages, in which it serves as a chaperone for delivery of nascent glucocerebrosidase polypeptide to the lysosomal compartment.Citation4–Citation6
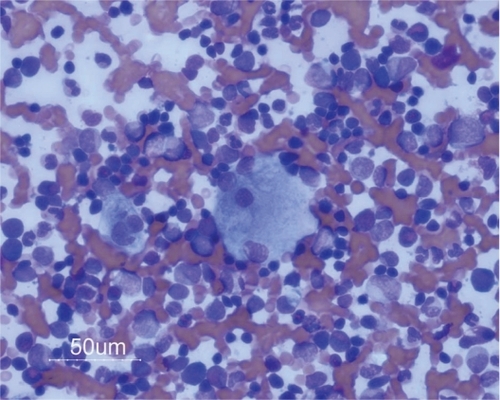
Gaucher disease most obviously affects cells of mononuclear phagocyte lineage in which prominent storage of undegraded lipids occurs (see ); mutations in the gene (GBA1) which encodes human β-glucocerebrosidase may disable the protein sufficiently to disturb its enzymatic function in other cell lineages, including those of the nervous system and the skin, which may become diseased.Citation3
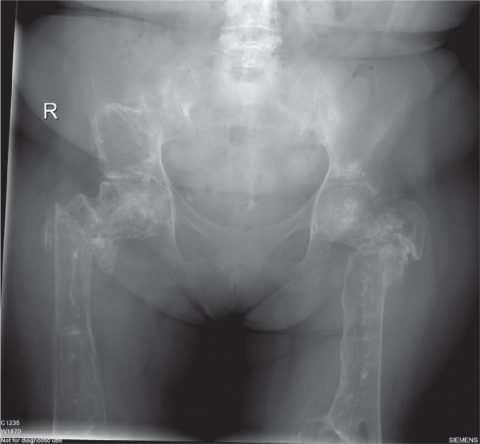
Figure 1 Pelvis, hips, and upper femora of adult Gaucher patient.
Clinical manifestations
Gaucher disease may occur at any age and in any human population.Citation3,Citation7–Citation9 Certain forms of the condition are over-represented in patients of Ashkenazi Jewish ancestry and in a small inbred population in Northern Sweden. For practical purposes, the condition has been classified into three principal ‘types’. The most frequent form of the condition (affecting ∼90% of patients and assigned as type I) is associated with subtotal deficiency of lysosomal acid β-glucocerebrosidase and manifestations principally in the viscera (principally liver and spleen, see ) as well as bone marrow, which are infiltrated by pathological macrophages (Gaucher cells, ) engorged with glucosylceramide and other sphingolipids. Many mutations responsible for the enzymatic deficiency have been identified in the glucocerebrosidase gene; those with severe effects on enzymatic activity are associated with various neurological manifestations and coinheritance of severe defects in the gene with almost complete lack of glucocerebrosidase, which is a rare cause of stillbirth with skeletal deformities and/or dehydration as a result of abnormal skin integrity (collodion babies). An acute form of neurological illness (for convenience classified as type II Gaucher disease but effectively a condition with neuronopathic features that are not slowly progressive, as in type III) is a very rare disorder causing death in the first years of life; characteristically, there is irritability, bulbar palsies, opisthotonus, and modest enlargement of the viscera.
Figure 2 Gaucher spleen >3 kg with large recent infarct.

Type I Gaucher disease is generally associated with prominent visceral and skeletal manifestations. Splenic pooling of formed elements of the blood leads to cytopenias with bleeding due to low platelet count, anemia, and risk of infection due to leukopenia. There may be massive enlargement of the liver and spleen () and occasionally severe infiltration of the lungs by pathological macrophages.
It is noteworthy that in the past, type I Gaucher disease was defined as being free of neurological manifestations, but increasingly it has been shown that patients develop an extrapyramidal disease resembling Parkinsonism in middle life, the cause of which is not understood. Moreover, it is now known that mutations in the human GBA1 gene, present in the heterozygous state in individuals of numerous ethnic origins and not hitherto identified as suffering from Gaucher disease, are the most prevalent genetic determinants of Parkinson’s disease so far identified in many populations.Citation10
Type I Gaucher patients have enlarged viscera which are infiltrated by alternatively activated macrophages which are typically found in the sinusoids of the spleen and replace the Kupffer cells of the liver. The macrophages are prominent in the bone marrow and contribute to acute episodes of osteonecrosis, particularly during growth. Necrosis of the marrow in proximity leads to impaired function of large joints, including the hip, knee, and shoulder. Other effects on the skeleton include local swellings (Gaucheromas) and destruction of bone in osteolytic lesions, as well as generalized demineralization and osteoporosis with an accompanying risk of fragility fractures (see ).
Figure 3 Pathological macrophages in bone marrow.
Less than 10% of patients with Gaucher disease develop a chronic so-called neuronopathic form, formerly termed type III Gaucher disease, with visual gaze palsies and other manifestations including myoclonic epilepsy and nerve deafness – all of which may be complicated by variable systemic involvement and at times prominent alveolar infiltration of the lungs.
Although Gaucher disease may declare itself at any age, in general, presentation in infancy and childhood carries a worse prognosis. There is a progressive deformity of the skeleton, enlargement of the viscera, and impaired function of organs, often accompanied by painful complications of skeletal disease. The condition shortens life, and even patients who present in late middle life ultimately may succumb. A rare complication of the condition is the development of hematological malignancies, most characteristically B-cell lymphomas and multiple myeloma, the pathogenesis of which is incompletely understood. Untreated Gaucher disease leads to progressive misery associated with bleeding, pallor, and anemia, together with visceral enlargement and bone pain; it is associated with disability and markedly reduced life quality, as well as survival.
Frequency
The birth frequency of Gaucher disease is ∼1:60,000 live births in the general population,Citation11 but genetic studies indicate a homozygote frequency of ∼1:950 Ashkenazi Jews, many of whom appear to remain asymptomatic.Citation12 In most countries, there is a marked discrepancy between the predicted and the observed prevalence: with a population of more than 60 million in the UK, only about 300 patients with Gaucher disease are known, giving an operational prevalence figure of about 1 in 200,000 – very much less than the predicted frequency at birth.Citation8 It can readily be seen that the disease is much less common than the threshold for designation as an orphan disorder in any country (eg, with a prevalence <1 in 2000 in Europe or <200,000 individuals in the United States); with <1 in 50,000 affected persons in the UK, the new term, ‘ultraorphan,’ has been suggested.
Diagnosis of Gaucher disease
The condition may be suspected in any patient with unexplained splenomegaly, particularly those of Ashkenazi Jewish origin.Citation3,Citation8,Citation12 Gaucher disease may also come to light as a result of investigations for pancytopenia or visceromegaly; thus Gaucher cells may be identified on tissue biopsy specimens, principally of bone marrow (during investigations for splenomegaly or cytopenias) or liver (during the course of investigations for hepatomegaly or abnormal liver-related biochemical tests). It is worth emphasizing that the presence of abnormal macrophages with the appearance of Gaucher cells is not sufficient for the diagnosis since these may be observed in other hematological conditions such as chronic myeloid leukemia, thalassemia, and multiple myeloma – the last named itself a rare complication of Gaucher disease. Specific diagnosis is, however, made by measuring acid β-glucosidase activity in fresh peripheral blood leukocytes, or occasionally by enzymatic analysis of fibroblasts cultured from skin biopsy specimens. Confirmation and better characterization of the condition may subsequently be afforded by molecular analysis of the human GBA1 gene, which encodes lysosomal glucocerebrosidase. The disorder may be suggested by a history of Perthés disease or as a result of radiological investigations which show bone deformity in the long bones due to a modeling defect, designated the Erlenmeyer flask abnormality, in the distal femur or proximal tibia.
Pathogenesis
In classical descriptions of the condition, although engorged macrophages represent the pathognomonic focus of Gaucher disease, the simple presence of storage material within these infiltrating cells provides an incomplete explanation of causation. Storage material accumulating within the macrophages accounts for a fraction of the massive bulk of the organs, which show macrophage hyperplasia and hypertrophy. While the pathological macrophages are accompanied by vascular changes and fibrosis, for example within the liver and spleen, they are not prominent in the brain in patients with neurological manifestations. More likely, abnormal storage material and other chemical changes within cells induce release of inflammatory factors, including chemokines and cytokines, which lead to the cascade of pathological changes.Citation3,Citation13–Citation20 Complex mechanisms, including abnormal calcium pooling within neural cell compartments, have been invoked, particularly in the neurological manifestations which characterize the severe Gaucher variants.Citation18,Citation19 Some authorities consider that the water-soluble lysolipid, glucosylsphingosine, has far-reaching signaling effects and may be responsible for cell death.Citation13,Citation16,Citation17 Quasipathophysiological concentrations of glucosylsphingosine affect the function of intracellular actin and induce a defect of cytokinesis generating a Gaucher-like phenotype on exposure of cultured macrophage cell lines to the molecule.Citation20
Therapeutics of Gaucher disease
Until the 1990s, no specific or curative treatments for most patients with Gaucher disease were available. Supportive and palliative measures include orthopedic interventions such as joint replacement surgery and occasional hepatic transplantation. Splenectomy was widely used to overcome the effects of hypersplenism in the blood, but carries with it immediate and long-term risks of overwhelming microbial infection;Citation21 splenectomy also contributes to the risk of osteonecrosis in Gaucher disease.Citation22
Cellular complementation
Given the visceral infiltration by pathological macrophages, which are of hematopoietic origin, bone marrow transplantation has been attempted in Gaucher disease.Citation23 Successful engraftment given before the development of irreversible skeletal and organ changes may correct the disease by replacing the defective macrophages with those of healthy donor origin. Children with the chronic neuronopathic form of Gaucher disease (type III), particularly those of Norrbottnian origin, had an improvement in their health but no apparent correction of the neurological disorder, despite a repopulation of microglia in the brain slowly by mononuclear cells of hematopoietic origin.Citation24,Citation25
Bone marrow and contemporary hematopoietic stem-cell transplantation is not in current general use for Gaucher disease, partly because of the shortage of ideal donors (human leukocyte antigen matched) and procedural risks, as well as the introduction of successful enzymatic augmentation which has superseded this treatment in many countries. The author treats one patient, the first male to receive allogeneic bone marrow transplantation in 1984 at the age of 11 years; homozygous for the L444P allele of the human glucocerebrosidase gene, the patient has type III disease with lateral gaze palsy and severe skeletal abnormalities following removal of a massive spleen aged 2 years. No longer confined to a wheelchair and with complete donor chimerism, the patient is now working independently as a farmer and drives a tractor unaided; he has no clinical or hematologic signs of Gaucher disease, which is inactive in the viscera and skeleton. From this case and a few others reported, it is clear that most, if not all, key aspects of the systemic disease are due to defective cells of mononuclear phagocytic lineage and thus may be corrected by supplying healthy exogenous hematopoietic stem cells or by transducing autologous stem cells with vectors for transferring the wild-type human glucocerebrosidase gene in order to restore the capacity of tissue macrophages to digest glycolipids normally.
Enzymatic augmentation
Soon after the enzymatic defect in Gaucher disease was identified in the 1960s, a long program of work into enzymatic replacement was initiated. At the time, studies on the hepatic uptake of plasma glycoproteins by Ashwell and Morell had been shown to be related to lectin-like activity of cellular receptors.Citation26 A further body of work showed that uptake of proteins by macrophages occurred through a surface receptor recognizing terminal mannose residues on protein ligands.Citation27 Early attempts to correct the biochemical abnormalities of Gaucher disease in patients after infusion of purified human glucocerebrosidase prepared from human placentae met with scant success, but it was later shown that the enzyme from human placentae contained enzyme molecules with complex carbohydrate chains, and mostly with terminal sialic acid residues.Citation28 In key experiments, it was later shown that sequential removal of sialic acid, galactose, and N-acetylglucosamine using exoglycosidases prepared from plant sources yielded a molecular form of the enzyme that was avidly taken up by Kupffer cells and other preparations of macrophages.Citation29
This finding formed the basis of further clinical trials of mannosylated glucocerebrosidase in Gaucher patients – one of whom, a small boy, showed a striking improvement in red-cell and platelet counts with a substantial reduction in spleen size.Citation30 A remarkable collaboration between a patient organization (the National Gaucher Foundation), the National Institutes of Health, where Dr. Roscoe Brady and his colleagues were working, and a newly formed company, the Genzyme Corporation, led to a clinical trial involving 12 patients with type I Gaucher disease. The participants were studied for 6 months during a course of infusions of modified human placental glucocerebrosidase given every 2 weeks. A large dose of the enzyme was administered (60 IU/kg of body weight every 2 weeks), and in the event, all the trial participants showed beneficial responses in terms of blood count and visceral parameters.Citation31 Mannose-terminated human placental glucocerebrosidase, later known as alglucerase (Ceredase®;Genzyme Corporation, Cambridge, MA), was licensed under the special regulations introduced through the orphan drug legislation in the United States. It was notable that the trial, which was an open-label trial without a control arm, showed clear benefit on spleen size in those patients studied.Citation31 As indicated above, since the splenic enlargement with hypersplenism is a central determinant of the illness in Gaucher disease and splenectomy can be avoided if the spleen size is decreased, this pivotal clinical trial clearly addressed an unmet clinical need. At the same time, the use of glucocerebrosidase derived from pooled human tissues (many thousands of human placentae were required for the production of sufficient Ceredase to treat a single Gaucher patient) posed considerable risk at a time when biological agents derived from human tissues were a known source of hepatitis viruses and HIV and were implicated in the transmission of the Creutzfeldt–Jakob agent. Nonetheless, numerous patients worldwide were treated with alglucerase under license, with salutary effects on quality of life, as well as improved blood counts, the regression of visceromegaly, and a decreased frequency of osteonecrosis episodes and reported bone pain.
Genetically engineered enzymatic augmentation
Approximately 5 years after the licensing of alglucerase, the Genzyme Corporation launched Cerezyme, imiglucerase – a recombinant human glucocerebrosidase expressed in genetically engineered Chinese hamster ovary cells. As with the purified placental product, this enzyme required further modification by exoglycosidases to expose glycan residues which mediate delivery to tissue macrophages utilizing the lectin-like properties of membrane mannose receptors.
Imiglucerase has now been given to more than 5000 patients worldwide and clearly reverses many of the manifestations of Gaucher disease, particularly those affecting the bone marrow and viscera.Citation32 In adults and children, there is a salutary increase in hemoglobin concentration, and white cell, and platelet counts, with a decrease in surrogate biomarkers of disease activity – certain macrophage-related proteins are released abundantly into the circulation in response to cellular storage.Citation33,Citation34
Tissue sampling, which is not routinely performed, demonstrates a reduction in the number of pathological macrophages infiltrating bone marrow and liver tissue. Although episodes of osteonecrosis become less frequent in patients receiving enzyme therapy and salutary improvements in bone mineralization density have been demonstrated in those with osteopenia and osteonecrosis, it is not surprising that those manifestations related to tissue and bone injury before treatment is instigated cannot be corrected.Citation35 While as a whole, the clinical evidence points strongly to disease-modifying properties of enzyme therapy in Gaucher disease, it is unclear as to whether it reduces the risk of B-cell malignancies and myeloma in patients with Gaucher disease. Moreover, any relationship of enzyme therapy to the development of the Parkinsonian complications of Gaucher disease has yet to be explored. Despite these reservations, enzyme replacement therapy for Gaucher disease with alglucerase and imiglucerase has been a signal therapeutic and hence commercial success, lifting the Genzyme Corporation to among the world’s largest biotechnology companies and feeding blockbuster revenues into its burgeoning fortune. In 2008, the total revenue of Genzyme was US$4.5 billion, of which sales of Cerezyme amounted to ∼US$1.2 billion. In Europe, Cerezyme was licensed under the newly introduced orphan medicinal products legislation for the chronic neuronopathic type III variant of Gaucher disease, where it also improves life quality, as well as the hematologic manifestations and visceral engorgement.Citation9,Citation36,Citation37
Biosimilar protein agents
The term ‘biosimilar’–otherwise known as ‘follow-on’ biologic agents–is used to describe approved new versions of innovative biopharmaceutical products following patent expiry. With respect to the current position of Cerezyme, the designation may be disputed in the industry. Although Cerezyme remains the standard care for the treatment of Gaucher disease and there is a burgeoning literature on its use over time in the mature phase of enzyme therapy,Citation35–Citation38 two emerging biosimilar agents, also based on the principle of macrophage targeting through the mannose lectin membrane receptor system, have been introduced. The first of these, velaglucerase-alfa (VPRIV®), was generated by a rival company, originally Transkaryotic Therapies, now taken over by Shire Human Genetic Therapies (Dublin, Ireland).Citation39 This agent is generated by gene activation of the endogenous human glucocerebrosidase gene in an immortalized human fibrosarcoma cell line. The engineered cells are cultured in a medium containing the powerful inhibitor kifunesine, which blocks the action of one of the processing glycosidases for glycoprotein biosynthesis, and as a result, a human glucocerebrosidase protein displaying terminal mannose sugars is produced. The other biosimilar agent in late-phase development is taliglucerase-alfa, an agent licensed by the Protalix company, based in Israel. Taliglucerase is produced as a recombinant glycoprotein expressed in genetically engineered plant cells.Citation40 To secure secretion through the vacuolar pathway, the protein is modified: it harbors additional amino acids, as well as xylose and other sugars in its intermediate glycan sequence. Clinical trial data, now fast emerging from these products, indicate therapeutic activity in type I Gaucher patients attributed also to targeted delivery and uptake by tissue macrophages.Citation39–Citation41
Disadvantages of enzyme replacement therapy
Contrary to expectations, hypersensitivity and immune reactions directed against the therapeutic proteins in type I Gaucher disease are very rare, and <1% of patients with this form of the condition manifest resistance to enzyme therapy. Not surprisingly, the neurological manifestations of Gaucher disease are not corrected by enzyme therapy – a failure attributed to the blood–brain barrier which is largely impermeable to proteins. However, in rural areas and undeveloped countries, the requirements for intravenous infusion pose difficulties for administration and delivery of treatment – often even the supply of sterile needles and infusion apparatus presents a challenge. In addition, the high intrinsic costs of biological and other therapies for Gaucher disease are discussed below. Despite its salutary effects and reversal of the hematopoietic factors of Gaucher disease, enzyme therapy has limitations and only a proportion of patients achieve their therapeutic goals.Citation38,Citation42 Enzyme therapy has no direct therapeutic effect on the neurological manifestations of Gaucher disease.Citation43–Citation45
Finally, it must be admitted that intravenous infusions are not the preferred means of therapy for many patients who, given the choice, would prefer an orally active agent to the perceived invasiveness, inconvenience, and discomfort and apparent ‘medicalization’ signified by an intravenous treatment.
Substrate depletion (inhibitor) therapy
As with enzymatic augmentation, this approach to therapy is based on sound biochemical principles; it moreover represents a telling example of scientific translation into clinical practice within a fiercely competitive orphan disease niche.
The concept of substrate depletion therapy in the glycosphingolipid diseases, of which Gaucher disease is an example, was introduced by Norman Radin and colleagues in the late 1970s,Citation46,Citation47 but it is based on a principle also adopted by Akira Endo and collaborators for the treatment of atherosclerosis due to hypercholesterolemia. Endo discovered and developed the class of fungal metabolites now known as statins used widely to inhibit cholesterol biosynthesis.Citation48
Since the inability to break down complex glycosphingolipids is the proximate cause of Gaucher disease and held to be the principal factor in its pathogenesis, inhibitors to decrease the biosynthesis of the substrate (glucosylceramide) should evince therapeutic benefit. Given that most patients have residual glucocerebrosidase activity, attenuating biosynthesis should allow this remaining enzymatic function to reduce steady-state concentrations of undegraded macromolecular substrate within lysosomes and, by rebalancing glycosphingolipid metabolism, ultimately correct the disease. The biochemical target for this stratagem in Gaucher disease is the first committed step for glycosphingolipid biosynthesis catalyzed by uridine diphosphate (UDP) glucosylceramide synthetase (UDP-glucose: N-acylsphingosine transferase).Citation47–Citation49 Two chemical classes of inhibitor are undergoing comprehensive therapeutic exploration: these are iminosugars derived from naturally occurring plant products and another class of compounds containing a pyrrolidine ring that serve as ceramide analogs.
Miglustat
The original compounds synthesized by Radin and colleagues were useful experimental inhibitors of the UDP-glucosylceramide synthase, but because of their appreciable cellular toxicity, they were not initially developed for further clinical application.Citation50 It was the iminosugars, in particular N-butyldeoxynojirimycin, previously explored for an unrelated application in HIV infection, which were identified for clinical development by Frances Platt and Terry Butters at the University of Oxford.Citation51,Citation52 They recognized that micromolar concentrations of N-butyldeoxynojirimycin inhibited the biosynthesis of glucosylceramide in cultures of a murine macrophage cell line treated with an irreversible inhibitor of acid-β-glucocerebrosidase, conduritol-β-epoxide. This agent induces lysosomal abnormalities, accompanied by an accumulation of glucosylceramide but coaddition of N-butyldeoxynojirimycin abrogated the lysosomal storage. Subsequent animal studies were conducted in knock-out mice lacking β-hexosaminidases, with accumulation of GM2 ganglioside in the brain.Citation53 A strain of these animals, the Sandhoff mouse, has a shortened life expectancy with neurological manifestations, accompanied by progressive storage of GM2 ganglioside throughout the central neuraxis. The administration of N-butyldeoxynojirimycin reduced ganglioside storage in peripheral organs and brain of these animals and extended their survival by ∼40%.Citation54
These preclinical studies, combined with earlier clinical trials in humans with HIV infection, stimulated the design of a clinical trial of N-butyldeoxynojirimycin (now known as miglustat or Zavesca®) in patients with type I Gaucher disease.Citation55 At a dose of 100 mg thrice daily, the agent reduced visceral enlargement and slowly improved hematologic parameters, as well as surrogate plasma biomarkers, in patients with type I Gaucher disease.Citation56–Citation58 An unwanted effect of the iminosugar treatment was diarrhea, caused by an inhibition of intestinal disaccharidase activity. Some patients also developed tremor and/or peripheral neuropathy, but the drug has been licensed in the United States and Europe as a second-line treatment for patients with mild to moderate type I Gaucher disease.Citation59
A recent study of its use in 28 patients with Gaucher disease who have been previously stabilized with enzyme therapy reported long-term findings with respect to organ size, blood counts, biomarkers, bone marrow infiltration, and safety, as well as tolerability. Assessments during routine clinic visits were carried out at 6, 12, 24, 36, and 48 months after initiating treatment with miglustat. Biomarkers improved up to 48 months after initiation of miglustat, while other parameters were reported to be stable. Miglustat was considered to have acceptable safety and tolerability and to be effective for the long-term maintenance of this group of patients with type I Gaucher disease who had previously received enzyme therapy.Citation60 While the authors of this report contend that their study represents ‘real-life’ clinical experience in the era after introduction of Ceredase and Cerezyme, the failure to distinguish the effects of miglustat from the course of type I Gaucher disease after long-term treatment (stabilization) with enzyme therapy or the confounding influence of patient selection, detracts from the scientific clarity of this study.
Since in some strains of mice the agent leads to a nongenotoxic sterility of males, strict contraception is advised and the drug is not licensed for use in children.Citation61–Citation63
With the potential for the small iminosugar molecule to penetrate the blood–brain barrier, a further trial was conducted in children with chronic neuronopathic Gaucher disease. However, this trial failed to meet its clinical end points and the drug currently is not recommended for neurological manifestations in Gaucher disease;Citation64 it is unknown as to whether or not it would be effective at higher tolerable doses in humans. Of interest miglustat Zavesca has recently received a license in Niemann–Pick disease type C, another lysosomal disease affecting the brain in which disturbed cholesterol trafficking to lysosomes causes secondary accumulation of glycosphingolipids in neurons.Citation65 Since there is no other effective treatment for this devastating neurodegenerative disorder, Zavesca delivers, in part at least, an unmet clinical need for patients and their families otherwise without hope. Zavesca was first developed by Oxford Glycosciences (Oxon, UK) and is now licensed for marketing under the orphan drug legislation by the Actelion company (Allschwil, Switzerland).
Eliglustat tartrate (Genz-112638)
James Shayman and colleagues in the University of Michigan and a former student of Norman Radin identified high-affinity inhibitors of the metabolic target of glycosphingolipid synthesis, UDP-glucyosylceramide synthase.Citation66 Homologues of the most potent inhibitors were generated as highly selective inhibitors of the enzyme, with inhibitory concentrations in the nanomolar range, but without appreciable cytotoxicity in culture. Cytotoxicity appears to be related to increased ceramide concentrations in cells, attributed to nonselective inhibitory effects of the prototype agents on a previously unknown ceramide transacylase activity. Intensive screening of isomers of a parent compound led to the generation of Genz-112638, recently developed as eliglustat tartrate by the Genzyme Corporation.Citation67 Medicinal chemists within the company invented the means for efficient enantioselective syntheses of the highly selective inhibitory isomers, which are held under patent.Citation68 The compound chosen for development is a structural analog of d-threo-ethylinedioxypheny l-2-palmitoylamino-3-pyrrilidino-propanol formulated as a salt of tartaric acid. The full chemical name of eliglustat tartrate is (1R, 2R)-Octanoic acid [2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide-l-tartaric acid salt.
This agent, as a free base, is metabolized by the cytochrome P450 system and in several cell lines has a powerful inhibitory action on UDP-glucose: N-acylsphingosine transferase with an IC50 of 24 nanomolar.Citation67 At low micromolar concentrations, it has no appreciable inhibitory action on intestinal sucrase and maltase activities or lysosomal acid β-glucocerebrosidase. Preclinical studies were conducted in a murine model of Gaucher disease (gbaD409V/nul), which develops occasional storage cells in viscera in association with lysosomal accumulation of glucosylceramide.Citation69 Eliglustat tartrate is a substrate for the P-glycoprotein multi-drug transporter and so does not accumulate in brain tissue of normal rodents. It was shown to have satisfactory preclinical toxicology in experimental animals.
Phase I studies have been conducted in 99 human subjects, with doses of 50–200 mg given twice daily inducing plasma concentrations of the agent within the predicted therapeutic range.Citation70 Although in preclinical studies the agent may have an effect on cardiac conduction intervals, this effect does not appear to be significant at therapeutic dosing levels. The drug is metabolized principally by the CYP2D6 cytochrome, thus careful monitoring will be prudent in patients taking other drugs known to interact with this metabolic pathway of degradation. Such drugs include paroxetine, ketoconazole, and rifampicin.
After satisfactory Phase I clinical trials in healthy human volunteers, Phase II clinical studies were undertaken, and information is available from patients having completed 2 years of therapy.Citation71,Citation72 These trials were undertaken in adults with type I Gaucher disease, for which the entry criteria required splenic enlargement of at least 10-fold normal, together with thrombocytopenia and/or anemia. The dose of drug was either started at 50 mg twice daily or with monitoring for pharmacokinetics adjusted to 100 mg twice daily to ensure that rapid metabolizers would have concentrations of the drug of ≈10 ng/mL. A composite primary efficacy end point, based on two of the three parameters (spleen volume, hemoglobin concentration, and platelet count), was used; furthermore, dual-energy X-ray absorptiometry studies with T1-weighted MRI of femurs were undertaken. There was extensive safety monitoring, particularly for untoward neurological effects and cardiac events.Citation72
Outcome measures
Of the 22 patients who completed the trial at 1 year, 20 attained the composite primary therapeutic end point, with improvements of at least two of the three abnormal values initially present. No bone crises occurred, and there were no changes in mobility, in bone pain, or on X-ray examination. There were, however, early signs of improvement in magnetic resonance imaging of bone marrow signal (dark marrow) at 2 years of treatment. Twenty of the original 26 patients had completed the therapy and most remain on an extended trial. Continuing improvement in spleen and liver volumes (the former decreased by a mean of 52%) with improvement in hemoglobin concentration and a rise in platelet counts have been observed. All these changes were accompanied by salutary improvements in surrogate biomarkers, including the chemokine CCL18-PARC and chitotriosidase activity. Of the 18 patients with abnormal dark signal independently identified on magnetic resonance imaging, six had improved by 1 year and an additional two patients had shown improvements by 2 years on the trial.Citation72 Further promising effects on bone mineralization density were seen with significant increases noted in most patients.
The safety profile and efficacy of eliglustat in patients with moderate Gaucher disease and the salutary effect on bone mineral density and abnormal bone marrow signals have prompted initiation of several multicenter Phase III studies. The first of these trials is a randomized, open-label study for adults with type I Gaucher disease, designed to compare the efficacy and safety of eliglustat tartrate with that of Cerezyme. Recruited patients should have received enzyme therapy for at least 3 years and have achieved clinical stability by reaching key therapeutic goals in terms of blood counts, visceral volumes, and bone status. The second trial is a randomized, blind, placebo-controlled study for patients with a confirmed diagnosis of type I Gaucher disease, who have not been treated for at least 12 months. A final trial has been registered, which will seek to compare the effects of one daily dosing of eliglustat tartrate with twice daily administration.
Pharmacological chaperone therapy
The pharmacological chaperone concept is based on the ability of small molecules to interact with mutant proteins that are misfolded and thus prevented from realizing their normal intracellular activity. Abnormal protein folding has been recognized as a common molecular mechanism in many inherited diseases and leads to premature degradation of the newly synthesized protein in the endoplasmic reticulum and also within the Golgi complex. In the case of lysosomal enzymes, the chaperone concept involves the binding of the agent to the active site of the mutant lysosomal protein, thus stabilizing it for delivery to its normal site of action in the acidic environment of the organelle. Chaperone molecules are thus often weak inhibitors which bind at neutral pH during biosynthesis of the enzyme and stabilize it for delivery to the lysosome; in the lysosome, the dramatic increase in hydrogen ion concentration is proposed to favor dissociation of the inhibitor, thus allowing restitution of enzyme function.Citation73
Another iminosugar, isofagomine, shows pharmacological chaperone activity directed toward glucocerebrosidase in fibroblasts cultured from type I Gaucher patients. This agent has been developed for Phase I/II clinical trials in Gaucher patients by the Amicus company (Cranbury, NJ). Although the drug was moderately well tolerated, apart from inducing conjunctivitis in some subjects, the first trial, with intermittent dosing as required for a putative pharmacological chaperone, showed disappointing therapeutic outcomes. The Amicus company continues with its trials, in particular with a similar iminosugar (1-deoxygalactonojirimycin) to investigate its possible therapeutic action on mutant α-galactosidase A in the related lysosomal disease, Anderson–Fabry disease.Citation73
Gene therapy
While in the context of enzyme therapy, which is safe and effective, the risks of bone marrow transplantation or hematopoietic stem-cell therapy render cellular complementation impractical for general application in most patients with Gaucher disease, the same stringencies might not be obtained for gene therapy using lentivirus-transduced autologous hematopoietic stem cells. If used for Gaucher disease, as reported for the neurodegenerative disease adrenoleukodystrophyCitation74 and currently in clinical trials for presymptomatic metachromatic leukodystrophy in infants and children, such an approach would have the advantage of being a one-off procedure requiring less powerful myelo-ablative conditioning and thus attractive for patients predicted to be at risk of severe disease and originating from countries where the availability, monitoring, or delivery of enzyme therapy is unsatisfactory.
Early clinical trials of macrophage-directed glucocerebrosidase using first-generation lentiviral gene transfer vectors in two centers were disappointing with very low transduction efficiencies and/or early shutoff of corrective gene expression in circulating monocytes; no clinical benefit was reported. These results were particularly disappointing since a bicistronic retroviral vector expressing human glucocerebrosidase and a human small cell surface antigen (CD24) as a selectable marker, under the control of the Moloney murine leukemia viral promoter, were used to transform CD34+ hematopoietic progenitors.Citation75 Latterly, there have been promising preclinical developments in the gene therapy of type I Gaucher disease from Stefan Karlsson and colleagues at the University of Lund, Sweden. This group was the first to generate a convincing inducible mammalian model of the disorder in the hematopoietic system and major viscera of genetically engineered mice;Citation76 they have further utilized this model as a test system to evaluate lentiviral gene therapy directed to autologous hematopoietic stem cells using busulfan for nonablative pretransplant conditioning without whole body irradiation. With this stratagem, they were able to demonstrate that stem-cell engraftment in the range of 1%–10% wild type confers clear benefit in this authentic disease model.Citation77
While there may be residual safety issues surrounding the use of lentiviral vectors directed to proliferating cellular compartments, the capacity permanently to correct the principal manifestations of the condition with a minor proportion of autologous stem cells corrected by gene transfer is a signal advance.Citation77 Recent studies on the use of third-generation self-inactivating (SIN) lentiviral vector derivatives in patients with adrenoleukodystrophy with random, rather than preferential, integration at sites with the potentiality for oncogenesis in the human genome are also critically important for issues of safety.Citation74,Citation75 However, in the recent clinical use of SIN lentiviral vectors to transduce autologous CD34+ hematopoietic stem cells in two boys with adrenoleukodystrophy by Cartier and colleagues, a complete myelo-ablative regimen was employed.
A further approach to the definitive treatment of non-neuronopathic Gaucher disease (type I) in effect combines gene therapy with systemic enzymatic complementation. In a stratagem using adeno-associated viral (AAV)-mediated gene therapy, McEachern and colleaguesCitation78 administered a recombinant AAV8 serotype vector harboring the human β-glucocerebrosidase gene under the control of a liver-selective promoter intravenously to D409V/null mice with features of Gaucher disease. The vector induced sustained hepatic secretion of the enzyme, which was sufficient to prevent accumulation of glucosylceramide and Gaucher cells in the liver, spleen, and lungs of young animals and had marked benefit on these parameters in older mice with established disease. The absence of antibodies on challenge indicated that the animals had been tolerized to the therapeutic protein. Such a strategy would also lend itself to the application of gene therapy to patients with Gaucher disease: it has the potential advantage of not requiring myelo-ablative therapy and ease of administration for what would be envisaged as a one-off procedure. A key requirement, however, would be sustained expression of the therapeutic gene in hepatocytes transduced by the rAAV vector – an issue that has yet to be overcome in relation to gene therapy for hemophilia.Citation79 Given the current state of knowledge and preclinical studies, credible clinical trials could soon be initiated; but the location of appropriate investigative centers and selection of patients will be of critical importance.
Discussion
Predictable and unexpected effects of orphan drug legislation
While the orphan drug legislation was designed to provide strong incentives for pharmaceutical development in neglected and rare diseases, the commercial rewards realized in a very rare (ultraorphan) condition, Gaucher disease, were unforeseen and unprecedented. The incentives have been keenly felt by several companies seeking similar trophies; they have sought also to position themselves in the field by acquisition.
Access and cost
There can be no doubt that the general outlook for patients with Gaucher disease and related lysosomal diseases for which enzymatic augmentation has been introduced (as well as initiatives involving small molecule inhibitors and chaperones) has improved immeasurably; these benefits extend only to those patients for whom insurance and national health care provision is available. Only a minority (≈10%) of patients with Gaucher disease worldwide are fortunate enough to have access to enzyme replacement therapy, principally because of the extreme cost of this ultraorphan treatment for each patient. To capitalize on its leading position and sustain a program of therapeutic development, Genzyme charges highly for products provided to only a few thousand patients internationally: the average cost to treat an adult Gaucher patient with enzyme therapy is of the order of £100,000 and in the early debulking phases of the illness, about £200,000 per annum.
Development costs
Although it is claimed that the introduction of a new biological drug may cost $500,000–$1,000,000,000, in the case of alglucerase (Ceredase) for Gaucher disease, the manufacturer reported spending less than $58M for development. However, we should remember that this was 20 years ago and the burden of costs was shared with academia. Given the prolonged marketing exclusivity awarded for the first introduction of an orphan agent of 7 and 10 years in the United States and Europe, respectively, the potential rewards for each corporate winner are high. While those developing biosimilar drugs must bide their time or demonstrate that their products have unequivocal therapeutic advantages, companies with an active discovery portfolio can steal an advantage if their drug has an innovative mode of action, as illustrated by the substrate inhibitors. Since it was first proposed, progress on gene therapy has been slow, even for monogenic diseases in which it clearly offers the potential for a definitive and specific cure. It is salutary to recall that more than two decades ago, the original scientific board of the corporation advised the management of Genzyme not to proceed with the development of enzyme therapy because gene therapy for the condition was so far advanced at the time. Perceptions of advantage and the real nature of competition in orphan diseases remain – as with all aspects of biotechnology – fraught with the foibles of human judgment and error.
Clearly, after a time, strong incentives also re-emerge for oncoming competitors with biosimilar protein agents, since by following the data of the innovator, development costs may be reduced to a scale of tens rather than hundreds of millions of dollars.
The value and safety implications of competition
We now also know that there is a previously unrecognized clinical advantage provided by alternative biological agents, for example velaglucerase-alfa and taliglucerase-alfa in Gaucher disease – a guaranteed safety of supply. The necessity of competing alternatives has emerged as a result of a catastrophic year-long shortage of Cerezyme, resulting from a vesivirus infection occurring in the dedicated bioreactor plant at the principal production facility of the Genzyme Corporation in Allston, Massachusetts, in June 2009.Citation80 At the time, although velaglucerase and taliglucerase were at an emerging stage of clinical development, their ready availability for several hundred patients in Europe and the United States has partially mitigated the crisis both for the patients and their treating physicians, as well as the prowess of the Genzyme Corporation. In the face of this crisis, many physicians and regulatory agencies worked with all the commercial partners to promote release of the new enzyme preparations through compassionate access programs before licensing to expedite the regulatory processes for licensing approval and to accelerate approval for distribution in the United States, Europe, and other regions, wherever possible.
The competitive niche
Biological treatments, such as enzymatic augmentation in lysosomal disorders, can be challenged by competitive small molecules, as has been convincingly demonstrated in Gaucher disease. The innovation of substrate depletion therapy with biosynthetic inhibitors of the principal glycolipids, which accumulate in macrophages, provides an attractive scientific and pharmaceutical alternative. Not only are such compounds more readily distributed and administered, but their mode of action is distinct and a compelling advantage in competition.
Other powerful advantages include the generally cheaper manufacturing costs of small molecular compounds, as compared with human enzyme preparations, which in the case of Gaucher disease require elaborate modification to ensure appropriate targeting to the affected tissues (macrophages) in vivo. While the Actelion drug, miglustat (Zavesca), is currently seen as a second-line agent for Gaucher disease,Citation59,Citation63 its inception clearly directed Genzyme to innovative departures, based on a distinct chemistry. Genzyme has made a large investment through numerous patents on the synthesis of its novel ceramide-like selective inhibitor of a novel biochemical target; and one wonders if it would have done so had a competing orally available agent with a novel mechanism of action not come to light.
In relation to pharmacological chaperone therapy, as yet in an undeveloped phase for clinical practice, it can be seen that the innovative drive and incentive for developing new therapies in Gaucher disease is pervasive and strong. Without it, much pharmaceutical development for needy patients with disorders otherwise lacking specific measures would not have occurred. While enzyme replacement therapy undoubtedly provides large measurable health gains for adults and may prevent disease if given sufficiently early in children, we must recognize that some accommodation is needed in our appreciation of pharmaceutical ‘miracles’. After all, if all rare diseases mandated such enthusiastic pharmaceutical interest, the costs of diverting health care resources to swell the revenues of pharmaceutical companies could not be sustained.
Pricing issues
The prices set by pharmaceutical companies for their drugs are arrived at by complex and obscure means, but are justified by the need to cover the costs of production and accompanying investment in the light of competing agents. The perception of health benefits is also an important factor in price setting. The need to generate profit is an unqualified but critical factor. In the case of orphan agents, the unmet need is construed as very high and unchallenged, for there is by definition no competition which would otherwise mitigate pricing. Although biological agents notionally would be expected to be very costly, the economy of scale and long-standing marketing exclusivity with, for example, the production of a genetically engineered protein from cell cultures quickly allows an astonishing profit margin to be achieved.
Issues of marketing exclusivity
Gaucher disease provides here another vivid example, not only of the triumph of Western capitalist principles of utility and progress linked to profit, but of a market exclusivity of another kind. For such are the prices of the drugs developed in Western democratic countries that the public health care systems of other populations do not even aspire to meet them. It will be recalled that Stalin commuted the salaries of doctors and other health care personnel greatly: even after the fall of Stalin and two decades after the collapse of the Communist system in Russia and post-Soviet states, exiguous salaries and investment have given rise to appalling state provision for health. At the same time, private medical services for party members and emerging plutocrats in Russia and many former Communist countries aspire to the best international standards of delivery and comfort. Under these circumstances, for many patients, access to expensive treatments such as Cerezyme for Gaucher disease is either denied or highly restricted.
Compassionate supply
The problem of access has been commendably addressed in the divisional locations of Genzyme in capitalist countries by the introduction of free compassionate use programs for severely ill Gaucher patients resident in communities where health care systems do not reimburse the costs of lifesaving therapy. However, this does not address the total need of Gaucher patients worldwide, for it is estimated that of the 5000 or so patients who have been treated with Cerezyme (until the recent shortage caused by vesivirus infection and shutdown of Genzyme’s Allston plantCitation80), this would represent a maximum of 10% of all symptomatic Gaucher patients.
Orphan drug pricing
While the high cost of manufacturing biological agents has the appearance of veracity, one should also expect the competing small molecules to be made available at costs that are substantially less than those for enzyme therapies. There is only one example for a direct comparison, namely Zavesca, at a cost of ∼$100,000 per year for an average adult. It is thus clear that incentive and shareholder revenue overshadow the costs of manufacture for nonbiological agents even in the orphan drug field.
The concept of ‘pile them high and sell them cheap’ has yet to emerge among the competitors of first-to-market orphan drugs, but where access remains difficult for many patients in less developed countries, this heretical idea arguably merits consideration.
Conclusion
Future considerations for orphan drug development
Market-driven economies reward value and should aspire to reward the value that health – a basic human necessity – brings to its citizens.
Some patients with rare diseases have now received welcome attention through the agency of orphan drug legislation, and in many cases, they gain real benefit as a result of pioneering humane initiatives. Nonetheless, the current model will need reviewing if it is to deliver value across the thousands of individually rare diseases in whole populations.Citation81
As illustrated by the remarkable example of Gaucher disease, orphan drug legislation can, in effect, promote vulnerable monopolies and also has other unintended consequences. Once licensed, beyond mandatory pharmacovigilance monitoring and fulfilment of postlicensing commitments, there is little incentive for the victorious company to invest in therapeutic research into the cognate disease. For such investment to occur, competition and the fear of loss of commercial primacy need to be felt, for only strong survival instincts and the need to demonstrate prowess will induce further scientific expenditure.
Orphan drug legislation is anticompetitive, but we now know that even this cannot guarantee the survival of any given drug, particularly a biologic agent like a therapeutic enzyme: there can be no immunity from unexpected manufacturing disasters. In the case of Gaucher disease, the catastrophic vesivirus infection that has all but stopped production of Cerezyme for most of the year from June 2009Citation80 (as well as Genzyme’s agent for Fabry disease, Fabrazyme®) leading to a reported 34% fall of Cerezyme sales to $793 million, while sales of Fabrazyme fell 13% to $494 million. The company is struggling now to recover its inventory while supplying as many of its global customers as possible: the catastrophe has brought home not simply the desirability but the absolute necessity of competition for the safe provision of alternative biosimilar agents.
Much has been learned from the modern miracle of Gaucher disease as an orphan disorder, but at the time of writing, we are entering a period of radical change. With a history of unresolved regulatory issues involving production at their impressive manufacturing plant after FDA inspections, the sudden appearance of a vesivirus infection at the Allston facility has exposed the Genzyme Corporation to intense scrutiny; the reputation of an inspirational leader in the field has been publicly questioned. Moreover, Genzyme has had to bear hostile bids in the ruthless commercial world of industrial takeovers – and in times when pharmaceutical giants are cutting their own research and development budgets in favour of acquiring successful biotech companies with more innovative approaches to rare diseases (‘niche-busting’). Although there can be no retreat, at a time of unparalleled opportunity and competitive interest in rare disorders, some form of societal reckoning to ensure sustainable innovation in the whole field – of which Gaucher disease is emblematic – is surely warranted.Citation1 Imaginative approaches will be needed to commute the prices of orphan drugs in a mature market, so that hand-in-hand therapeutic discovery and development can be maintained in the long term at the current unprecedented level of productivity and clinical benefit.
Acknowledgements
Research into Gaucher disease from the author’s group is supported in part by the European Union, 7th Frame Programme ‘Euclyd – a European Consortium for Lysosomal Storage Diseases’ health F2/2008 grant agreement 201678 and by the Cambridge Biomedical Research Centre of the UK National Institute of Health Research of the Department of Health. The group has been previously supported by the Gauchers Association UK.
I sincerely thank Elizabeth Chatters for excellent secretarial assistance and Liz Morris, Patrick Deegan, Jane Tindall, Naomi Wright, Joan Grantham, and many other colleagues for their expert advice and loyal service in the care of innumerable patients with Gaucher disease attending our National center for lysosomal disorders at Addenbrooke’s hospital, supported by the National Commissioning Group of the National Health Service (England).
Disclosure
The author reports no conflicts of interest in this work in so far as neither he nor any member of his family has stocks or direct financial interests in any company as mandated by his election to current board membership of the European Working Group for the study of Gaucher disease (honorary vice-chairman, 1993–2010). The author receives lecturing and conference fees from Actelion, Genzyme, and Shire Human Genetic Therapies. His laboratory has received unrestricted research support from Genzyme and Shire for scientific projects unrelated to Gaucher disease. The author has provided occasional advice to Actelion, Genzyme, and Shire in relation to marketing authorization of their products and the conduct of clinical trials, as well as to regulatory agencies (EMEA and FDA) concerning cognate products of these companies and the treatment of Gaucher disease; similarly, he has advised Amicus and Protalix but without charge. The author is an investigator in clinical trials of treatments for Gaucher disease supplied by Actelion, Genzyme, Shire, and Protalix; he accepted investigator status for trials with Amicus, but the trial was closed before enrolment. No professional fees are paid to the author in respect of his status as clinical investigator and institutional involvement with trials conducted at Cambridge University NHS Foundation Hospitals Trust (based at Addenbrooke’s hospital).
References
- FuchsVRNew priorities for future biomedical innovationsN Engl J Med2010363870470620818873
- BradyROKanferJNShapiroDMetabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher’s diseaseBiochem Biophys Res Commun19651822122514282020
- BeutlerEGrabowskiGAGaucher diseaseScriverCRBeaudetALSlyWSValleDThe Metabolic and Molecular Basis of Inherited DiseaseIII8th edNew York, NYMcGraw-Hill200136353668
- ReczekDSchwakeMSchröderJLIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of β-glucocerebrosidaseCell2007131477078318022370
- BalreiraAGasparPCaiolaDA nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndromeHum Mol Genet200817142238224318424452
- BerkovicSFDibbensLMOshlackAArray-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosisAm J Hum Genet200882367368418308289
- ZimranAKayAGelbartTGaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patientsMedicine (Baltimore)19927163373531435229
- CoxTMSchofieldJPGaucher’s disease: clinical features and natural historyBaillieres Clin Haematol19971046576899497857
- EriksonAGaucher disease – Norrbottnian type (III). Neuropaediatric and neurobiological aspects of clinical patterns and treatmentActa Paediatr Scand Suppl1986326S1S42
- BultronGKacenaKPearsonDThe risk of Parkinson’s disease in type 1 Gaucher diseaseJ Inherit Metab Dis201033216717320177787
- MeiklePJHopwoodJJClagueAECareyWFPrevalence of lysosomal storage disordersJAMA199928132492549918480
- BeutlerENguyenNJHennebergerMWGaucher disease: gene frequencies in the Ashkenazi Jewish populationAm J Hum Genet199352185888434610
- CoxTMGaucher disease: understanding the molecular pathogenesis of sphingolipidosesJ Inherit Metab Dis200124Suppl 210612111758671
- AertsJMHollakCEPlasma and metabolic abnormalities in Gaucher’s diseaseBaillieres Clin Haematol19971046917099497858
- MoranMTSchofieldJPHaymanARShuGPYoungECoxTMPathologic gene expression in Gaucher disease: up-regulation of cysteine proteinases including osteoclastic cathepsin KBlood20009651969197810961902
- JmoudiakMFutermanAHGaucher disease: pathological mechanisms and modern managementBr J Haematol2005129217818815813845
- NilssonOSvennerholmLAccumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher diseaseJ Neurochem19823937097187097276
- PelledDTrajkovic-BodennecSLloyd-EvansESidranskyESchiffmannRFutermanAHEnhanced calcium release in the acute neuronopathic form of Gaucher diseaseNeurobiol Dis2005181838815649698
- FutermanAHvan MeerGThe cell biology of lysosomal storage disordersNat Rev Mol Cell Biol20045755456515232573
- ImD-SHeiseCENguyenTO’DowdBFLynchKRIdentification of a molecular target of psychosine and its role in globoid cell formationJ Cell Biol20011532420434
- CoxTMAertsJMBelmatougNManagement of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoringJ Inherit Metab Dis200831331933618509745
- DeeganPBPavlovaEVTindallJOsseous manifestations of adult Gaucher disease in the era of enzyme replacement therapyMedicine (Baltimore)2011901In press.
- HobbsJRJonesKHShawPJLindsayIHancockMBeneficial effect of pre-transplant splenectomy on displacement bone marrow transplantation for Gaucher’s syndromeLancet198718542111111152883444
- RingdenOGrothCGEriksonAGranqvistSMånssonJESparrelidETen years’ experience of bone marrow transplantation for Gaucher diseaseTransplantation19955968648707701581
- HoogerbruggePMBrouwerOFBordigoniPAllogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow TransplantationLancet19953458962139814027760610
- AshwellGMorellAGThe role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteinsAdv Enzymol Relat Areas Mol Biol197441991284609051
- StahlPDRodmanJSMillerMJSchlesingerPHEvidence for receptor-mediated binding of glycoproteins, glycoconjugates, and lysosomal glycosidases by alveolar macrophagesProc Natl Acad Sci U S A197875313991403274729
- BradyROEnzyme replacement for lysosomal diseasesAnn Rev Med20065728329616409150
- FurbishFSSteerCJKrettNLBarrangerJAUptake and distribution of placental glucocerebrosidase in rat hepatic cells and effects of sequential deglycosylationBiochim Biophys Acta198167344254346784774
- BartonNWFurbishFSMurrayGJGarfieldMBradyROTherapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher diseaseProc Natl Acad Sci U S A1990875191319162308952
- BartonNWBradyRODambrosiaJMReplacement therapy for inherited enzyme deficiency – macrophage-targeted glucocerebrosidase for Gaucher’s diseaseN Engl J Med199132421146414702023606
- GrabowskiGABartonNWPastoresGEnzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sourcesAnn Intern Med1995122133397985893
- WeinrebNJCharrowJAnderssonHCEffectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher registryAm J Med2002113211211912133749
- AnderssonHKaplanPKacenaKYeeJEight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1Pediatrics200812261182119019047232
- SimsKBPastoresGMWeinrebNJImprovement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort studyClin Genet200873543044018312448
- OnoHFujiwaraMItoKUedaHMizoguchiNSakuraNNeurological features in Gaucher’s disease during enzyme replacement therapyActa Paediatr200190222923111236058
- EriksonAForsbergHNilssonMAströmMMånssonJETen years’ experience of enzyme infusion therapy of Norrbottnian (type 3) Gaucher diseaseActa Paediatr200695331231716497642
- WeinrebNTaylorJCoxTYeeJvom DahlSA benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imigluceraseAm J Hematol2008831289089518819093
- ZimranAAltarescuGPhilipsMPhase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experienceBlood2010115234651465620299511
- ShaaltielYBartfeldDHashmueliSProduction of glucocerebrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher’s disease using a plant cell systemPlant Biotechnol J20075557959017524049
- AviezerDBrill-AlmonEShaaltielYA plant-derived recombinant human glucocerebrosidase enzyme – a preclinical and phase I investigationPLoS One200943e4792 Epub 2009 Mar 11.19277123
- PastoresGMWeinrebWJAertsHTherapeutic goals in the treatment of Gaucher diseaseSemin Hematol2004414 Suppl 541415468045
- SchiffmannRHeyesMPAertsJMProspective study of neurological responses to treatment with macrophage-targeted glucocerebrosidase in patients with type 3 Gaucher’s diseaseAnn Neurol19974246136219382473
- DaviesEHEriksonACollin-HistedTMengelETylki-SzymanskaAVellodiAOutcome of type III Gaucher disease on enzyme replacement therapy: review of 55 casesJ Inherit Metab Dis200730693594217994286
- EriksonARemaining problems in the management of patients with Gaucher diseaseJ Inherit Metab Dis200124Suppl 2122126 discussion 87–88.11758672
- VunnanRRRadinNSAnalogs of ceramide that inhibit glucocerebroside synthetase in mouse brainChem Phys Lipids19802632652786445239
- RadinNSTreatment of Gaucher disease with an enzyme inhibitorGlycoconj J19961321531578737239
- EndoAA gift from nature: the birth of the statinsNat Med200814101050105218841147
- InokuchiJRadinNSPreparation of the active isomer of 1-phenyl-2-decanoylamino-3-morpholino-1-propanol, inhibitor of murine glucocerebroside synthetaseJ Lipid Res19872855655712955067
- AbeARadinNSShaymanJAStructural and stereochemical studies of potent inhibitors of glucosylceramide synthase and tumor cell growthJ Lipid Res19953636116217775872
- PlattFMNeisesGRDwekRAButtersTN-butyldeoxnojirimycin is a novel inhibitor of glycolipid biosynthesisJ Biol Chem199426911836283658132559
- ButtersTDvan den BroekLAGMFleetGWJMolecular requirements of imino sugars for the selective control of N-linked glycosylation and glycosphingolipid biosynthesisTetrahedron Asymmetry2000111113124
- PlattFMNeisesGRReinkensmeierGPrevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycinScience199727653114284319103204
- JeyakumarMButtersTDCortina-BorjaMDelayed symptom onset and increased life expectancy in Sandhoff mice treated with N-butyldeoxynojirimycinProc Natl Acad Sci U S A199996116388639310339597
- CoxTLachmannRHollakCNovel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesisLancet200035592141481148510801168
- ElsteinDHollakCAertsJMSustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher diseaseJ Inherit Metab Dis200427675776615505381
- HeitnerRElsteinDAertsJWeelySZimranALow-dose N-butyldeoxynojirimycin (OGT 918) for type I Gaucher diseaseBlood Cells Mol Dis200228212713312064906
- PastoresGMBarnettNLKolodnyEHAn open-label, noncomparative study of miglustat in type 1 Gaucher disease: efficacy and tolerability over 24 months of treatmentClin Ther20052781215122716199246
- CoxTMAertsJMAndriaGThe role of the iminosugar N-butyldeoxynojirimycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease: a position statement. Advisory Council to the European Working Group on Gaucher DiseaseJ Inherit Metab Dis200326651352614605497
- GiraldoPAlfonsoPAtutxaKReal-world clinical experience with long-term miglustat maintenance therapy in type 1 Gaucher disease: the ZAGAL projectHaematologica200994121771177519608672
- van der SpoelACJeyakumarMButtersTDReversible infertility in male mice after oral administration of alkylated imino sugars: a nonhormonal approach to male contraceptionProc Natl Acad Sci U S A20029926171731717812477936
- SuganumaRWaldenCMButtersTDAlkylated imino sugars, reversible male infertility-inducing agents, do not affect the genetic integrity of male mouse germ cells during short-term treatment despite induction of sperm deformitiesBiol Reprod200572480581315576825
- WeinrebNJBarrangerJACharrowJGrabowskiGAMankinHJMistryPGuidance on the use of miglustat for treating patients with type 1 Gaucher diseaseAm J Hematol200580322322916247743
- SchiffmannRFitzgibbonEJHarrisCRandomized, controlled trial of miglustat in Gaucher’s disease type 3Ann Neurol200864551452219067373
- WraithJEVecchioDJacklinEMiglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trialMol Genet Metab201099435135720045366
- LeeLAbeAShaymanJAImproved inhibitors of glucosylceramide synthaseJ Biol Chem199927421146621466910329660
- McEachernKAFungJKomarnitskySA specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher diseaseMol Genet Metab200791325926717509920
- HirthBHSiegelCSynthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors. United States patent US 68558302005215
- MarshallJMcEachernKAChuangWLImproved management of lysosomal glucosylceramide levels in a mouse model of type 1 Gaucher disease using enzyme and substrate reduction therapyJ Inherit Metab Dis201033328128920336375
- PeterschmittMJBurkeABlanksteinLSafety, tolerability, and pharmacokinetics of eliglustat tartrate (Genz-112638) after single doses, multiple doses, and food in healthy volunteersJ Clin Pharmacol In press 2010. MS Reference: JCP-09-Nov-437.R1.
- LukinaEWatmanNArreguinEAA phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1Blood2010116689389920439622
- LukinaEWatmanNAvila ArreguinEImprovement in hematological, visceral and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz 112638) treatment: two-year results of a phase 2 studyBlood2010816 [Epub ahead of print]
- ParentiGTreating lysosomal storage diseases with pharmacological chaperones: from concept to clinicsEMBO Mol Med20091526827920049730
- CartierNHacein-Bey-AbinaSBartholomaeCCHematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophyScience2009326595481882319892975
- MigitaMMedinJAPawliukRSelection of transduced CD34+ progenitors and enzymatic correction of cells from Gaucher patients, with bicistronic vectorsProc Natl Acad Sci U S A1995922612075120798618847
- EnquistIBNilssonEOokaAEffective cell and gene therapy in a murine model of Gaucher diseaseProc Natl Acad Sci U S A200610337138191382416954197
- EnquistIBNilssonEMånssonJEEhingerMRichterJKarlssonSSuccessful low-risk hematopoietic cell therapy in a mouse model of type 1 Gaucher diseaseStem Cells200927374475219056909
- McEachernKANietupskiJBChuangWLAAV8-mediated expression of glucocerebrosidase ameliorates the storage pathology in the visceral organs of a mouse model of Gaucher diseaseJ Gene Med20068671972916528760
- HighKAThe Jeremiah Metzger lecture: gene therapy for inherited disorders: from Christmas disease to Leber’s amaurosisTrans Am Clin Climatol Assoc200912033135919768188
- HollakCEvom DahlSAertsJMForce majeure: therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher diseaseBlood Cells Mol Dis2010441414719804996
- RoosJCHyryHICoxTMOrphan drug pricing my warrant a competition law investigationBr Med J2010341c64711084108621081598