
Abstract
Non-alcoholic fatty liver disease (NAFLD) is a clinicopathological change characterized by the accumulation of triglycerides in hepatocytes and has frequently been associated with obesity, type 2 diabetes mellitus, hyperlipidemia, and insulin resistance. It is an increasingly recognized condition that has become the most common liver disorder in developed countries, affecting over one-third of the population and is associated with increased cardiovascular- and liver-related mortality. NAFLD is a spectrum of disorders, beginning as simple steatosis. In about 15% of all NAFLD cases, simple steatosis can evolve into non-alcoholic steatohepatitis, a medley of inflammation, hepatocellular injury, and fibrosis, often resulting in cirrhosis and even hepatocellular cancer. However, the molecular mechanism underlying NAFLD progression is not completely understood. Its pathogenesis has often been interpreted by the “double-hit” hypothesis. The primary insult or the “first hit” includes lipid accumulation in the liver, followed by a “second hit” in which proinflammatory mediators induce inflammation, hepatocellular injury, and fibrosis. Nowadays, a more complex model suggests that fatty acids (FAs) and their metabolites may be the true lipotoxic agents that contribute to NAFLD progression; a multiple parallel hits hypothesis has also been suggested. In NAFLD patients, insulin resistance leads to hepatic steatosis via multiple mechanisms. Despite the excess hepatic accumulation of FAs in NAFLD, it has been described that not only de novo FA synthesis is increased, but FAs are also taken up from the serum. Furthermore, a decrease in mitochondrial FA oxidation and secretion of very-low-density lipoproteins has been reported. This review discusses the molecular mechanisms that underlie the pathophysiological changes of hepatic lipid metabolism that contribute to NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a major public health issue due to its high prevalence worldwide, and ranges widely from 11% to 46%,Citation1–Citation3 and has potentially serious sequelae.Citation4 The prevalence increases to 58% in overweight individuals and can be as high as 98% in non-diabetic obese individuals.Citation5
NAFLD is an inclusive term that takes in a spectrum of liver pathologies from simple steatosis (SS) to non-alcoholic steatohepatitis (NASH). NASH involves hepatocellular injury and inflammation of the liver.Citation6
Whereas SS is characterized by a relatively favorable clinical course, NASH much more frequently progresses to cirrhosis and hepatocellular carcinoma.Citation7,Citation8 NAFLD should be suspected in individuals who are either obese, diabetic, or have metabolic syndrome.Citation9
Moreover, NAFLD is considered a hepatic manifestation of metabolic syndrome and a risk factor for type 2 diabetes mellitus, dyslipidemia, and hypertension.Citation10,Citation11 The majority of patients with NAFLD are asymptomatic and the disease may be detected via routine blood tests showing elevated liver enzymes or when an ultrasound is performed for various reasons and detects liver steatosis. Secondary causes of hepatic steatosis or elevated liver enzymes, such as excess alcohol consumption, medications, toxins, lipodystrophy, autoimmune and inflammatory diseases, nutrition (malnutrition, total parenteral nutrition, severe weight loss, and refeeding syndrome), viral hepatitis, and metabolic liver disease should be excluded by reviewing the patient’s history and proper investigation.Citation9,Citation12
Although it is still not possible to diagnose NAFLD based solely on blood work, elevated transaminases can be used as a first step.Citation13 An aspartate aminotransferase–alanine aminotransferase ratio <1 is also seen in NAFLDCitation14 and supports NASH. However, it is important to note that patients with normal transaminases and liver steatosis on imaging may also have NASH.Citation15 Ultrasonography is a noninvasive tool that is used in the detection of liver steatosis. Other imaging techniques such as computed tomography and nuclear magnetic resonance imaging can also detect liver steatosis, but neither of these more expensive techniques provide more information than ultrasonography,Citation16,Citation17 except for fat quantification.Citation18 Diagnosis for NASH is confirmed when a liver biopsy shows the presence of perilobular inflammation, or the presence of hepatocyte ballooning, Mallory’s hyaline, and acidophil bodies with or without fibrosis. Noninvasive tests such as Fatty Liver Index, NAFLD fibrosis score, FibroMeter, and FibroscanCitation19 may suggest the presence of NASH by detecting fibrosis. Research is ongoing to assess surrogate markers for NASH such as CK18, but this remains experimental.Citation20,Citation21
Regarding the management of NAFLD, weight management through improvements in diet and increased physical activity can help to improve liver histology as well as delay disease progression.Citation22–Citation24 Lifestyle interventions may not be effective in certain cases, and thus other approaches must be considered. Pharmacological treatment has been studied in this population, specifically insulin-sensitizing agents (metformin and thiazolidinediones [TZDs]); however, there are conflicting results. Clinical studies could not demonstrate the effectiveness of metformin in the treatment of NAFLD.Citation25 On the other hand, TZDs that are peroxisomal proliferator-activated receptor γ (PPARγ) agonists promote hepatic fatty acid (FA) oxidation and decrease hepatic lipogenesis.Citation26,Citation27 In NAFLD patients, TZDs have been shown to decrease hepatic fat and decrease cellular injury. However, discontinuing TZD therapy resulted in NASH recurrence and long-term use of TZDs can result in medical complications such as edema, congestive heart failure, osteoporosis, and weight gain.Citation28,Citation29 The use of statins in NAFLD patients with dyslipidemia can improve liver function tests,Citation30 as well as steatosis.Citation31 Furthermore, statins seem to be safe in NAFLD/NASH patients with dyslipidemia.Citation32 However, there is a lack of evidence for the use of statins in the treatment of NASH patients without dyslipidemia.Citation33 Further research is necessary to document the effect of other strategies, such as bariatric surgery, antioxidants, and fish oil in NAFLD.
Because there are currently no effective therapies for NAFLD apart from weight loss, ongoing research efforts are focused on understanding the underlying pathobiology of hepatic steatosis with the intention of identifying novel therapeutic targets. In this sense, this review analyses some of the molecular mechanisms that underlie the pathophysiological changes of hepatic lipid metabolism in NAFLD: the contribution of lipid metabolism, the influence of inflammation, and the role of lipotoxicity and cannabinoid receptors in NAFLD.
Contribution of lipid metabolism to NAFLD
The liver plays a major role in lipid metabolism, importing free FAs (FFAs) and manufacturing, storing, and exporting lipids; derangements in any of these processes can lead to the development of NAFLD.Citation34 FAs are involved in many important cellular events, such as synthesis of cellular membranes, energy storage, and intracellular signaling pathways. However, chronically elevated FFAs can disturb diverse metabolic pathways and induce insulin resistance (IR) in many organs. Hepatic fat accumulation has been strongly associated with IR.Citation35,Citation36 IR in the peripheral adipose tissue enhances lipolysis and increases the delivery of adipose-derived FFAs to the liver. In particular, obesity increases tumor necrosis factor α (TNFα) production in adipocytes, facilitates adipocyte IR, and increases lipolysis rate.Citation37 Thus, the circulating pool of FFAs is increased in obese individuals and accounts for the majority of liver lipids in NAFLD.Citation38
Under physiological conditions, triglyceride (TG) synthesis is stimulated to dispose of the excess of FFAs. The TGs can then be stored as lipid droplets within hepatocytes or secreted into the blood as very-low-density lipoprotein (VLDL).Citation39 Rodent studies have shown that the mechanisms leading to the excessive accumulation of hepatic TGs are associated with an increased supply of FFAs from peripheral adipose tissue to the liver and an enhanced de novo lipid synthesis via the lipogenic pathway. Conversely, liver disposal via β-oxidation and VLDL export are moderately affected.Citation40 At the cellular level, defects in the insulin signaling pathways contribute to the increase of FFA flux in the liver, which in turn activates a series of signaling cascades and leads to the phosphorylation of several substrates.Citation41 Despite TGs being the main lipids stored in the liver of patients with NAFLD, large epidemiological studies suggest that they might exert protective functions. TG synthesis seems to be an adaptive, beneficial response in situations where hepatocytes are exposed to potentially toxic TG metabolites.Citation42–Citation44 FFAs and cholesterol, especially when accumulated in the mitochondria, are considered the “aggressive” lipids leading to TNFα-mediated liver damage and reactive oxygen species (ROS) formation.Citation45,Citation46 These lipids could also be present in a non-steatotic liver and act as early “inflammatory” hits, leading to the whole spectrum of NAFLD pathologies. The concept of lipotoxicity and involved lipid species has been introduced: abundant FAs cause lipotoxicity via the induction of ROS release, which causes inflammation, apoptosis, and thus, the progression to NASH and fibrogenesis.Citation46–Citation48
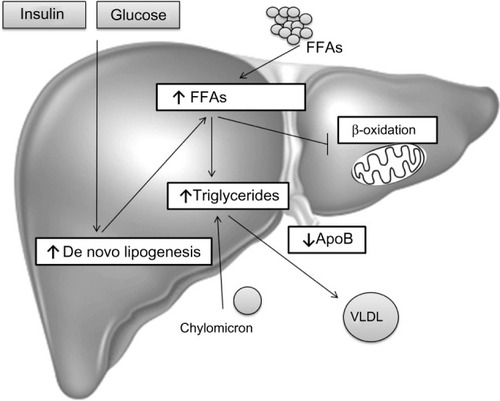
In summary, TG accumulation in the cytoplasm of hepatocytes, as the hallmark of NAFLD, arises from an imbalance between lipid acquisition (FA uptake and de novo lipogenesis) and removal (mitochondrial FA oxidation and export as a component of VLDL particles) and accompanies multiple pathophysiological mechanisms in NASH (). In order to control the progression of NAFLD, it is important to understand the regulatory mechanisms of lipid accumulation in the human liver.
Figure 1 Hepatic steatosis.
Abbreviations: ApoB, apolipoprotein B; FFAs, free fatty acids; FA, fatty acid; NAFLD, non-alcoholic fatty liver disease; VLDL, very-low-density lipoprotein.
Hepatic FA uptake
One of the sources for hepatic FAs is FFA recruitment from the blood stream. FFAs are derived from lipolysis in adipocytes, which usually occurs in the fasting state, promoted by catecholamines, natriuretic peptides, and glucagon, and are usually repressed by insulin.Citation49 However, the IR state (obesity, metabolic syndrome) goes along with increased adipocyte lipolysis, leading to abundant FFAs in the plasma pool independently from the nutritional status.Citation50 FFAs are then taken up by the hepatocytes in a facilitated fashion rather than by passive processes.Citation51 FA uptake into the liver contributes to the steady balance of hepatic TGs, as well as the pathogenesis of NAFLD. The rate of FA uptake from plasma into cells depends on the FA concentration in the plasma and the hepatocellular capacity for FA uptake, which also depends on the number and activity of transporter proteins on the sinusoidal plasma membrane of the hepatocyte. The main plasma membrane transporters for FFAs are FA transporter protein (FATP), caveolins, FA translocase (FAT)/CD36, and FA-binding protein (FABP).Citation52–Citation56
FATP
Six FATP isoforms have been identified in mammalian cells, which contain a common motif for FA uptake and fatty acyl-CoA synthetase function.Citation57 Of these isoforms, FATP2 and FATP5 are highly expressed in the liver, and are utilized as major FATPs for the normal physiological context. In mouse hepatocytes, adenovirus-mediated knockdown of FATP2 or genetic deletion of FATP5 significantly decreases the rates of FA uptake.Citation58 Indeed, FATP5 knockout mice have shown resistance to diet-induced obesity and hepatic TG accumulation.Citation58 In humans, a promoter polymorphism in the liver-specific FATP5 is associated with features of the metabolic syndrome and steatosis.Citation59
Caveolins
Caveolins consist of three protein family members termed caveolins 1, 2, and 3. They are found in the membrane structures called caveolae, which are important for protein trafficking and the formation of lipid droplets. Caveolin 1 knockout mice exhibited lower TG accumulation in the liver and showed resistance to diet-induced obesity, showing the importance of this protein in TG synthesis.Citation60 Some authors suggest there is an involvement of caveolin 1 in abnormal lipogenesis and mitochondrial function typical of steatotic hepatocytes in NAFLD.Citation61
FAT/CD36
It is well-known that FFAs are taken up into cells by passive diffusion and by protein-mediated mechanisms involving a number of FA transporters, of which FAT/CD36 is the best characterized. FAT/CD36 is expressed in a wide variety of cells including macrophages, adipocytes, myocytes, enterocytes, and hepatocytes. This transmembrane protein plays an important role in facilitating the uptake and intracellular trafficking of FFAs, as well as esterification into TGs in heart and skeletal muscle cells; this function is largely dependent on its translocation from intracellular depots to the plasma membrane. Insulin, muscular contractions, and the transcription factor Forkhead box protein O1 (FoxO1) induce FAT/CD36 translocation and enhance FFA uptake.Citation62
Hepatic FAT/CD36 expression is normally weak, but its expression is enhanced in rodents with fatty liver.Citation63 Moreover, some authors have demonstrated that FAT/CD36 mRNA levels increase concomitantly with hepatic TG content in different animal models of liver steatosis.Citation64,Citation65 Further studies have shown that FAT/CD36 is a common target gene of liver X receptor (LXR), pregnane X receptor, and PPARγ in promoting hepatic steatosis in a murine model.Citation66 However, little is known about the significance of FAT/CD36 in human liver diseases. In morbidly obese patients with NAFLD, Greco et al showed that hepatic FAT/CD36 mRNA levels were positively related to liver fat contentCitation67 and Bechmann et al found a significant correlation between hepatic FAT/CD36 mRNA and apoptosis in patients with NASH.Citation68 Other authors have described that hepatic FAT/CD36 upregulation is significantly associated with IR, hyperinsulinemia, and increased steatosis in patients with NASH.Citation62
FABPs
The FABPs are a group of molecules that coordinate inflammatory and metabolic responses in cells.Citation69 These proteins are a family of 14- to 15-kDa proteins that bind with high affinity to hydrophobic ligands such as saturated and unsaturated long-chain FAs (LCFAs).Citation70 Two isoforms of FABPs, aP2 (FABP4) and mal1 (FABP5) are the isoforms coexpressed in adipocytes and macrophages.Citation71 The expression of these FABP isoforms is controlled transcriptionally during adipocyte differentiation and is regulated by PPARγ agonists, insulin, and FAs. The functions of cytoplasmic FABPs include enhancement of FFA solubility and transport to specific enzymes and cellular compartments (to the mitochondria and peroxisomes for oxidation; to the endoplasmic reticulum [ER] for reesterification; into lipid droplets for storage; or to the nucleus for gene expression regulation).Citation71,Citation72 Disruption or pharmacological blockade of FABP4 protects mice from dyslipidemia, atherosclerosis, IR, and fatty liver in the context of either a high-fat diet or genetically induced obesity.Citation73 The definitive biology and function of FABPs in human physiology and disease are still not fully clarified.Citation69,Citation73 Few studies have assessed the involvement of hepatic FABP4 expression in NAFLD. Greco et al and Taskinen et al have described FABP4 as being upregulated in subjects with high liver fat content.Citation67,Citation74 The expression of FABP4 and FABP5 in the liver was correlated with hepatic fatty infiltration in NAFLD patients.Citation75
Recent studies have also suggested that hepatic FA uptake via FATPs can be a novel therapeutic strategy for NAFLD. Adenovirus-mediated knockdown of FATP2 or FATP5 reduced hepatic TG accumulation in high-fat fed mice.Citation76,Citation77 Moreover, both deoxycholic and ursodeoxycholic acid have shown promise as inhibitors of FATP5-mediated FA uptake, suggesting that they may improve hepatic steatosis in high-fat fed mice.Citation78
PPARγ
PPARγ is a master transcriptional regulator of adipogenesis and plays an important role in the process of lipid storage.Citation79 Thus, PPARα and PPARγ have opposing functions in the regulation of fat metabolism; PPARα promotes utilization, while activation of PPARγ promotes storage. Indeed, as increased PPARγ expression has been found in steatotic livers, it has been suggested that the role of PPARγ in the activation of lipogenic genes may contribute to the development of steatosis. Nevertheless, several studies have shown that PPARγ overexpression can prevent the progression of hepatic steatosis in murine models, and treatment with the PPARγ agonist rosiglitazone has been shown to have similar effects. The protective effects of PPARγ could be due to higher insulin sensitivity in adipose tissue and skeletal muscle leading to a reduction in FFA deposition in the liver. Adiponectin has also been shown to be increased by PPARγ, which also contributes to insulin sensitivity as well as upregulating PPARα expression, which leads to further hepatic FA oxidation. Furthermore, PPARγ expression has been shown to have anti-inflammatory and anti-fibrotic effects in stellate cells, macrophages, and epithelial cells. Westerbacka et al have described that PPARγ was overexpressed in the fatty liver of obese human subjects.Citation75
Activation of PPARγ in adipose tissue has been proposed to promote the relocalization and storage of fat in adipose tissue, protecting peripheral tissues from lipotoxicity.
Regarding this, the TZDs have proven to be effective drugs for improving insulin sensitivity and treating type 2 diabetes. Moreover, pioglitazone and rosiglitazone are highly effective in improving NAFLD outcomes in patients.Citation80 Unfortunately, the clinical use of TZDs against NAFLD has been hampered by side effects.
De novo lipogenesis
The process in which the liver synthesizes endogenous FAs is called de novo lipogenesis. This includes de novo synthesis of FAs through a complex cytosolic polymerization in which glucose is converted to acetyl-CoA by glycolysis and the oxidation of pyruvate. Acetyl-CoA carboxylase (ACC1) then converts acetyl-CoA into malonyl-CoA. Finally, FA synthase (FAS) catalyzes the formation of palmitic acid from malonyl-CoA and acetyl-CoA.Citation81–Citation83 Depending on the metabolic state, FAs are then processed to TGs and stored or rapidly metabolized.
Dietary fats are packed in chylomicrons and hydrolyzed, releasing FAs of which approximately 20% are delivered to the liver.Citation7 In the fasting state, a decline of insulin levels stimulates adipocyte TG hydrolase, thereby releasing FFAs that are transported to the liver. In the liver, FFAs derived from peripheral tissue, endogenous synthesis, or diet, can be used for: 1) energy and ketone body production via mitochondrial β-oxidation; 2) sterified and stored as TGs in lipid droplets; or 3) packaged with apolipoprotein B into VLDL that is secreted into the circulation.Citation83,Citation84 In NAFLD patients, enhanced acquisition of FAs through uptake and de novo lipogenesis are not compensated by FA oxidation or production of VLDL particles ().
The rate of de novo lipogenesis is regulated primarily at the transcriptional level. Several nuclear transcription factors are involved such as LxRα, sterol regulatory element-binding protein 1c (SREBP1c), carbohydrate-responsive element-binding protein (ChREBP), and farnesoid X receptor (FxR); and enzymes (ACC1, FAS, and steroyl CoA desaturase 1 [SCD1]). Postprandially, plasma glucose and insulin levels rise, which promote activation of lipogenesis through the activation of ChREBP and SREBP1c, respectively.Citation34,Citation85 In humans, NAFLD has been associated with increased hepatic expression of several genes involved in de novo lipogenesis.Citation86,Citation87
LXRs
LXRs are ligand-activated transcription factors that belong to the nuclear receptor (NR) superfamily.Citation88 There are two LXR isoforms termed α and β. LxRα is mainly expressed in the liver, adipose tissue, and intestine, whereas LxRβ is ubiquitously expressed.Citation89 In addition to modulating cholesterol metabolism, LXRs have been characterized as major regulators of hepatic FA biosynthesis.Citation90 A major function of LxRα in the liver is the stimulation of de novo lipogenesis, through the induction of SREBP1c, ACC1, FAS, and SCD1 ().Citation91–Citation93
Figure 2 Transcriptional control of lipogenesis and glycolysis.
Abbreviations: ACC, acetyl-CoA carboxylase; ChREBP, carbohydrate-responsive element-binding protein; FA, fatty acid; FAS, fatty acid synthase; FFAs, free fatty acids; FxR, farnesoid X receptor; G6PC, glucose 6-phosphatase; GCKR, glucokinase regulatory protein; LXR, liver X receptor; MUFA, monosaturated fatty acids; PPARα, peroxisomal proliferator-activated receptor alpha; SCD1, steroyl CoA desaturase 1; SFA, saturated fatty acids; SREBP1c, sterol regulatory element-binding protein 1c; TG, triglyceride.
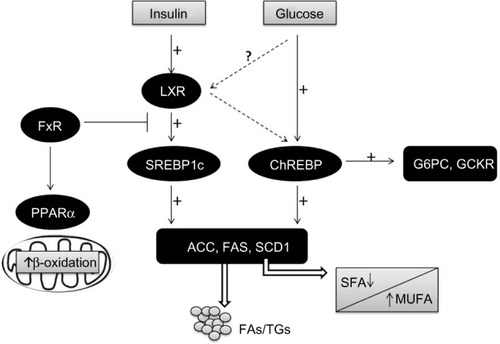
Several authors have described an enhanced expression of LxRα and SREBP1c in NAFLD.Citation87,Citation94,Citation95
SREBP1c
SREBPs are a family of membrane-bound transcription factors. SREBPs are synthesized as 125 kD precursors embedded in the ER. Proteolytic cleavage then allows the accumulation of active SREBP in the nucleus.
There are different SREBP isoforms: SREBP1c and SREBP2 are expressed in the liver, while SREBP1a is expressed only at very low levels in the liver of adult mice, rats, and humans.Citation96 SREBP1c, the predominant isoform in the liver, preferentially affects the transcription of genes that regulate de novo lipid synthesis, although SREBP2 regulates genes involved in cholesterol biosynthesis and metabolism. The SREBP1a isoform, despite its very low levels in the liver, transactivates both lipogenic and cholesterol genes.Citation97
To date, the main regulation demonstrated for SREBP1c is at the transcriptional level. SREBP1c transcription is induced by two quite disparate stimuli: insulin, a hormone released in response to carbohydrate intake and leading to a parallel increase in both the membrane-bound precursor and the mature nuclear form, and LxRα, a transcription factor that acts as a cholesterol sensor.Citation92,Citation93,Citation98–Citation100 In response to feeding, SREBP1c binds to its lipogenic genes, such as ACC1, FAS, and SCD1, and to its own gene, thereby stimulating hepatic lipogenesis ().Citation96,Citation101–Citation105
Different authors have described an enhanced expression of SREBP1c and LxRα in NAFLD.Citation87,Citation94,Citation95 However, Nagaya et al demonstrated the downregulation of the hepatic SREBP1c-mediated lipogenic pathway in advanced NASH patients; SREBP1c mRNA levels were inversely correlated with the fibrosis stage.Citation106 These discrepancies might be explained by differences in the cohort of studied patients. For example, Higuchi et alCitation94 included normal weight patients with NAFLD and Lima-Cabello et alCitation95 included patients with NAFLD and with steatosis related to chronic hepatitis C virus infection in mildly overweight men and women. Moreover, Higuchi et al did not evaluate either histological findings nor protein levels or intracellular localization of SREBP1c.Citation94
ChREBP
De novo lipogenesis is regulated by glucose and insulin signaling pathways in response to dietary carbohydrate intake to induce glycolytic and lipogenic gene expression. SREBP1c has emerged as a major mediator of insulin action on lipogenic genes. However, SREBP1c activity alone is not sufficient for the stimulation of glycolytic and lipogenic gene expression.Citation107,Citation108 Over recent years, it has been reported that the liver transcription factor ChREBP is required for the induction of glycolytic gene expression by glucose and that it acts together with SREBP1c to stimulate lipogenic genes.Citation109–Citation111 Interestingly, ChREBP was also identified as a direct target of LXRs, which are an important regulator of the lipogenic pathway through the transcriptional control of SREBP1c, ACC1, FAS, and SCD1.Citation112–Citation117 Oxysterols are known as ligands of LXRs, but glucose was also shown to activate LXRs and to induce their target genes, including ChREBP ().Citation107,Citation118
Postprandial hyperglycemia raises the hepatic concentrations of phosphorylated intermediates, causing activation of ChREBP, which binds to the promoter of its target genes as a heterotetramer with its ubiquitously expressed partner Max-like protein X (Mlx). ChREBP target genes include not only enzymes of glycolysis and lipogenesis that predispose to hepatic steatosis, but also glucose 6-phosphatase (G6PC), which catalyzes the final reaction in glucose production, and glucokinase regulatory protein (GCKR), which inhibits hepatic glucose uptake.Citation119,Citation120 Transcriptional induction of G6PC and GCKR manifests as hepatic glucose intolerance or IR.Citation121 Studies using a dominant negative variant of Mlx identified target genes of ChREBP–Mlx that promote hepatic glucose intolerance when overexpressed.Citation120
Study results of the role and impact of ChREBP in glucose and lipid metabolism have been confusing. Global ChREBP deficiency in C57BL/6J mice results in IR.Citation122 On the other hand, ChREBP deficiencyCitation123,Citation124 or expression of a dominant negative Mlx isoformCitation125 in an obese ob/ob background decreases hepatic steatosis and other related metabolic alterations, including IR. Benhamed et alCitation126 hypothesized that these opposite phenotypes in these two murine models may reside in the fact that ChREBP controls both glycolysis and lipogenesis, and that the beneficial effect of ChREBP deficiency may only be apparent in the context of lipid overload. The authors showed that mice overexpressing ChREBP, on a standard diet, remained insulin sensitive, despite increased lipogenesis resulting in hepatic steatosis. However, mice that overexpress ChREBP, on a high-fat diet, showed normal insulin levels and improved insulin signaling and glucose tolerance compared with controls, despite having greater hepatic steatosis. This effect seems to be mediated by the fact that ChREBP modifies the monounsaturated FAs/saturated FAs (MUFA/SFA) balance in favor of MUFA, by stimulating SCD1 activity. Taken together, these results demonstrated that increasing ChREBP, by buffering detrimental FAs and favoring lipid partitioning, can dissociate hepatic steatosis from IR, with beneficial effects on both glucose and lipid metabolism. Interestingly, ChREBP expression in liver biopsies from patients with NASH was higher when steatosis was greater than 50% and lower in the presence of severe IR,Citation126 sup porting this conclusion.
Furthermore, because insulin induces enzymes of lipogenesis by activation of SREBP1c and represses G6PC through other transcriptional regulators, a mechanism of “selective IR” has been proposed to explain the simultaneous elevation of hepatic glucose production (or G6PC expression) and lipogenesis in human type 2 diabetes or models of IR.Citation127
FxR
The FxR is a member of the NR superfamily and a receptor for bile acids. FxR activation leads to alterations in pathways involved in energy metabolism. It is mainly expressed in the liver, intestine, kidneys, and the adrenal glands, with less expression in adipose tissue and heart.Citation128–Citation130 FxR has emerged as a master regulator of lipid and glucose homeostasis in the liver and of inflammatory processes at hepatic and extrahepatic sites. Also, a number of synthetic FxR agonists are being used for the treatment of different hepatic and metabolic disorders, resulting in a lower inflammatory and fibrogenic process.Citation131,Citation132
The generation of FxR knockout mice showed a clear role for FxR as the master regulation of bile acid homeostasis.Citation133,Citation134 However, FxR knockout mice also revealed elevated levels of cholesterol and TGs in both the plasma and liver, suggesting a key role for FxR lipid metabolism as well. In fact, it was demonstrated that FxR needs to be activated in order to reduce the expression of SREBP1c.Citation135 More recently, in addition to bile acid and lipid metabolism, it has been shown that FxR also plays an important role in glucose metabolism, improving insulin sensitivity and glucose tolerance in a diabetic mice model.Citation136,Citation137
Regarding its role in lipid metabolism, the majority of literature seems to point to the fact that FxR activation is beneficial in situations of excess, such as obesity and diabetes. FxR activation seems to reduce TGs levels by: 1) reducing FA synthesis in the liver, through the reduction of SREBP1c and LxR expression;Citation138 2) inducing the expression of PPARα, which promotes FFA catabolism via β-oxidation; 3) increasing TG clearance; and 4) increasing adipose tissue storage and altering adipokine patterns ().Citation139,Citation140
Another hepatic protective mechanism of FxR activation has been shown to be maintenance of gut integrity against gut-derived endotoxins through the induction of antibacterial factors such as angiogenin, inducible NO synthase, and interleukin-18 (IL18).Citation131,Citation132
Patients with NAFLD have lower protein and mRNA FxR levels, which has been attributed to higher TG synthesis and induced expression of SREBP1c and LxRα.Citation138
ACC1 and FAS
In the process of FA synthesis, ACC1 converts acetyl-CoA, an essential substrate of FAs, to malonyl-CoA. FAS then utilizes both acetyl-CoA and malonyl-CoA to form palmitic acid (C16:0). Both are highly regulated by a transcriptional factor, SREBP1c, and play important roles in the energy metabolism of FAs. They are currently considered an attractive target for regulating the human diseases of obesity, diabetes, cancer, and cardiovascular complications. Dorn et al found that FAS expression was impaired in SS, while the absence of SS in hepatic inflammation did not affect FAS expression.Citation141 In agreement with Dorn et al, several authors have described an enhanced expression of FAS in NAFLD.Citation142 These authors have also described that ACC1 mRNA expression was higher in NAFLD. In support of increased FA synthesis in NAFLD, Morgan et al found that ACC1 and FAS mRNA expression were significantly higher in high-fat mice.Citation143 All these findings suggest that ACC1 and FAS might be a new diagnostic marker or therapeutic target for NAFLD.
FoxO1
FoxO1 is a transcription factor with an important role not only in glycogenolysis and gluconeogenesis, but also in lipid metabolism.
With regard to lipid metabolism, liver-specific transgenic expression of active FoxO1 induces the expression of genes involved in lipid transport and decreases the expression of important genes for glycolysis and lipid/sterol synthesis, resulting in lower postprandial TG concentrations compared to in wild-type mice.Citation144 However, using a similar murine model, another group observed enhanced lipogenesis and liver steatosis.Citation145 Similarly, adenoviral delivery of an active FoxO1 variant to the liver results in lipogenesis, hepatic steatosis, and reduced FA oxidation. These increases in lipogenesis result from a feedback loop that enhances insulin signaling, thereby modulating lipid metabolism through SREBP1c in a FoxO1-independent manner.Citation146
FoxO1 not only inhibits SREBP1c expression but also suppresses the expression of genes directly involved in FA synthesis, including FAS and adenosine triphosphate (ATP) citrate lyase.Citation144
With regard to glucose metabolism, under fasting conditions, the liver provides energy by releasing glucose into the bloodstream. Initially, this results from the breakdown of liver glycogen stores (glycogenolysis), whereas with prolonged fasting, the primary source of glucose is gluconeogenesis. Studies with adenoviral vectors in isolated hepatocytes confirm that FoxO1 stimulates the expression of gluconeogenic genes and suppresses the expression of genes involved in glycolysis, the shunt pathway, and lipogenesis, including glucokinase and SREBP1c. Taken together, these results indicate that FoxO1 proteins promote hepatic glucose production through multiple mechanisms and contribute to the regulation of other important metabolic pathways in adapting to fasting and feeding in the liver, including glycolysis, the pentose phosphate shunt, and lipogenic and sterol synthetic pathways.Citation144
Chronic expression of an active FoxO1 mutant in the liver leads to increased expression of genes involved in gluconeogenesis, resulting in elevated plasma glucose and insulin levels, which are not able to maintain normal glycemia.Citation144 Valenti et al suggest that increased FoxO1 activity may play a role in the pathogenesis of hepatic IR associated with NAFLD.Citation147
Reduction of FoxO1 in both liver and white adipose tissue using an antisense oligonucleotide-mediated approach improves glucose tolerance and both hepatic and peripheral insulin action in mice with diet-induced obesity.Citation148 Consistent with these studies, FoxO1 haploinsufficiency is able to rescue the loss of insulin sensitivity in insulin receptor–haploinsufficient mice partly by reducing the hepatic expression of gluconeogenic genes.Citation149
FA oxidation
Oxidation of FAs occurs within the mitochondria, peroxisomes, and the ER. It facilitates the degradation of activated FAs to acetyl-CoA. FAs are activated by acyl-CoA-synthetase to acyl-CoA in the cytosol, which is indispensable for enabling FAs to cross membranes and enter organelles. Short- and medium-chain FAs pass the mitochondrial membrane without activation. However, activated LCFAs are shuttled across the membrane via carnitine palmitoyltransferase-1 (CPT1). Malonyl-CoA, an early intermediate of de novo lipogenesis, is an inhibitor of CPT1. In the fed state, FA oxidation is inhibited and de novo lipogenesis promoted, allowing for storage and distribution of lipids. In general, short-, medium-, and long-chain FAs are oxidized within mitochondria (β-oxidation), while toxic, very-long-chain FAs are oxidized within peroxisomes. In diabetes or FA overload, cytochrome P450 (CYP4A)-dependent ω-oxidation of LCFAs occurs in the ER and induces ROS and lipid peroxidation. During the process of β-oxidation, electrons are indirectly donated to the electron transport chain to drive ATP synthesis. Acetyl-CoA can be further processed via the tricarboxylic acid cycle, or in the case of FA abundance, be converted into ketone bodies. PPARα and insulin signaling are again involved in the regulation of FA oxidation and the formation of ketone bodies via transcriptional regulation of mitochondrial 3-hydroxy-3-methylglutaryl (HMG)-CoA synthase.Citation82
PPARα
In the liver, PPARα plays a pivotal role in FA metabolism by upregulating the expression of numerous genes involved in mitochondrial FA and peroxisome FA oxidation, as well as numerous other aspects of FA metabolism in the cell.Citation150 As a consequence, activation of PPARα can prevent and decrease hepatic fat storage.Citation151–Citation154 When PPARα sensing is inefficient, overnight or prolonged fasting leads to severe hepatic steatosis, as seen in PPAR-α−/− mice.Citation155,Citation156 PPARα−/− mice fail to upregulate FA oxidation systems in the liver and cannot oxidize the influxed FAs, and thus develop severe hepatic steatosis. PPARα−/− mice also develop severe steatohepatitis when maintained on a diet deficient in methionine and choline.Citation153,Citation157,Citation158 Also of importance is that administering PPARα agonists to rats not only prevents the development of methionine- and choline-deficient diet-induced steatohepatitis by preventing intrahepatic lipid and lipoperoxide accumulation, but also reverses hepatic fibrosis by decreasing the expression of fibrotic markers and reducing the number of stellate cells.Citation153,Citation157–Citation159 The efficacy of these agonists in the treatment of NAFLD in human subjects has not yet been studied in depth. From the available data, no definitive conclusion can be made on the efficacy of PPARα agonists on NAFLD due to study limitations, such as small sample size, incomplete data, and the use of agonists in combination with other strategies.Citation160
Besides governing metabolic processes, PPARα also regulates inflammatory processes, mainly by inhibiting inflammatory gene expression. Hepatic PPARα activation has been repeatedly shown to reduce hepatic inflammation elicited by acute exposure to cytokines and other compounds.Citation161–Citation165
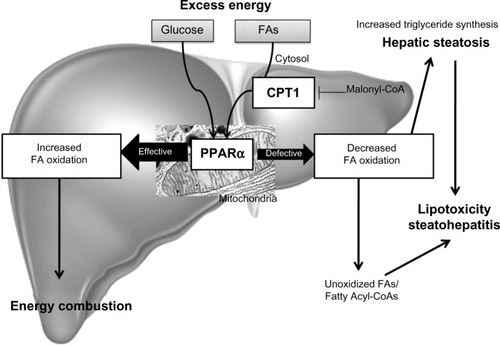
In conclusion, PPARα activation plays a role in the modulation of hepatic steatosis due to its effects: upregulation of FA oxidation systems and the ensuing burning of energy, reduction in the toxicity of FAs, and its anti-inflammatory effect ().Citation153,Citation155,Citation156,Citation158
Figure 3 Fatty acid oxidation.
Abbreviations: CPT1, carnitine palmitoytransferase-1; FA, fatty acid; PPARα, peroxisomal proliferator-activated receptor alpha.
CPT1
CPT1 is a regulatory enzyme in the mitochondria that transfers FAs from the cytosol to the mitochondria prior to β-oxidation (). Inhibition of CPT1 has been shown to prevent IR induced by a high-fat diet, partly due to a reduction in some of the deleterious intermediates generated by incomplete FA oxidation and partly to a shift toward increased glucose oxidation for energy production.Citation166 Kohjima et al showed that CPT1 expression in humans is reduced by 50% in NAFLD compared with that in the normal liver.Citation142
Inflammation and NAFLD
It is well-known that the balance between pro- and anti-inflammatory acting cytokines is fundamental in the control of systemic and hepatic insulin action, and as a consequence, in the development of NAFLD. IR is an important feature of NAFLD and is caused by a variety of factors, including soluble mediators derived from adipose tissue and/or immune cells: the adipocytokines ().Citation167
Figure 4 Cytokines and NAFLD.
Abbreviations: ADIPOR2, adiponectin receptor type 2; IL6, interleukin-6; NAFLD, non-alcoholic fatty liver disease; TNFα, tumor necrosis factor alpha; TNFR, tumor necrosis factor receptor; IL6R, interleukin-6 receptor.
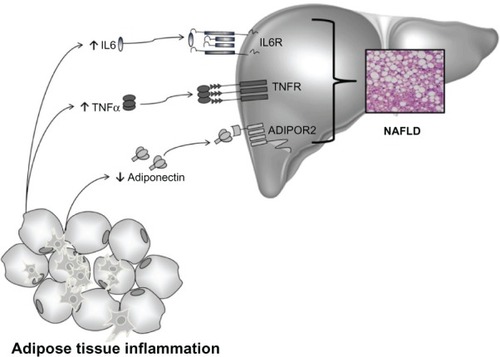
Adiponectin
Adiponectin, one of the major products of adipocytes, is a prototypic anti-inflammatory and anti-diabetic adipocytokine, the actions of which are mainly exerted by the activation of adenosine monophosphate (AMP)-activated kinase and PPARα. Adiponectin has two specific receptors: adiponectin receptor type 1 and 2 (ADIPOR1 and 2). ADIPOR1 is widely expressed, whereas ADIPOR2 can be mainly observed in the liver.Citation168 Serum levels of adiponectin are lower in individuals with obesity, type 2 diabetes, and in conditions of IR,Citation169 whereas adiponectin synthesis is induced by weight loss and PPARγ activation by its ligands, TZDs.Citation28 In general, studies have suggested that adiponectin exerts anti-inflammatory effects, stimulates secretion of anti-inflammatory cytokines such as IL10 or IL1 receptor antagonist (IL1Ra), blocks nuclear factor κB (NF-κB) activation, and inhibits the release of TNFα, IL6, and chemokines.Citation170,Citation171
The liver is not a relevant source of circulating adiponectin, but it is a major target organ for many of its effects. In mice with alcoholic and non-alcoholic fatty liver disease, administering recombinant adiponectin ameliorated necroinflammation and steatosis, partly via inhibition of the hepatic production of TNFα and the decrease in plasma concentration of this proinflammatory cytokine.Citation172
In humans, adiponectin serum levels were lower in patients with NASH in comparison to matched controls and to patients with SS, independently of IR or the waist–hip ratio. IR and low adiponectin serum levels were associated with increased steatosis and necroinflammation, but not with severe fibrosis, which was predicted only by IR.Citation173 Another study has shown that morbidly obese patients with IR undergoing bariatric surgery have lower mRNA adiponectin expression in adipose tissue and lower serum levels of adiponectin than those without IR. This low expression of adiponectin may predispose patients to the progressive form of NAFLD or to NASH.Citation174
Low mRNA expression of adiponectin and ADIPOR2 was found in the liver of patients with NASH compared with those with SS. Moreover, ADIPOR2 expression was inversely related to alanine aminotransferase and the fibrosis stage.Citation175 More recently, Moschen et al demonstrated in a prospective study that rapid weight loss after bariatric surgery results in a significant improvement of both histological and biochemical liver parameters, which is accompanied by an increase of adiponectin serum levels, as well as hepatic mRNA adiponectin expression.Citation176
Altogether, there is now strong evidence that circulating adiponectin levels are lower in obesity and related human disorders, including NAFLD.Citation176,Citation177
TNFα and IL6
TNFα and IL6 are two important proinflammatory adipocytokines and the expression of both is hugely increased in the fat cells of obese human subjects and patients with IR.Citation178,Citation179 In patients with severe obesity, the mRNA expression of IL6 and TNFα is clearer in adipose compared to liver tissue.Citation180
TNFα was identified more than two decades ago as the first inflammatory molecule linked with IR.Citation181 Higher serum levels of TNFα and soluble TNFα receptor 2 (TNFR2) have been found in patients with NASH compared with healthy subjects and these differences were independent of higher IR. However, no significant differences in TNFα and TNFR2 were found between SS and NASH patients.Citation173
Enhanced TNFα hepatic expression was recently demonstrated in a group of obese patients with NAFLD. Crespo et al reported increased hepatic expression of TNFα and TNFR2 in patients with NASH compared to patients with SS. In these patients, more advanced fibrosis was also accompanied by the increased hepatic expression of TNFα.Citation182 In line with these results, TNFα plasma levels have been shown to correlate positively with the grade of liver fibrosis assessed by ultrasound-guided liver biopsy in patients with advanced stages of NAFLD.Citation183
Furthermore, certain TNFα polygenetic polymorphisms have been found to have higher IR indices, a higher prevalence of impaired glucose tolerance, and higher susceptibility to the development of NAFLD and NASH.Citation184,Citation185
IL6 is a pleiotropic cytokine expressed in many inflammatory cells in response to different types of stimuli, regulating a number of biological processes including IR and the regulation of inflammation. It is known to be the main stimulating factor for hepatocyte synthesis and the secretion of C-reactive protein in humans,Citation186 and for this reason, it has been proposed as a potential mediator leading to NAFLD. However, the true mechanisms driving IL6 induced NAFLD remain unclear.
Preliminary studies have found that IL6 plays a protective role in liver fibrosis by promoting hepatocyte proliferation and by protecting against oxidative stress and mitochondrial dysfunction.Citation187 On the other hand, Wieckowska et al demonstrated markedly increased IL6 expression in the liver of patients with NASH compared to those with SS or normal liver. Hepatic IL6 expression also correlated positively with the severity of inflammation and fibrosis. IL6 plasma levels that were measured in parallel in this study correlated well with liver IL6 expression.Citation6 In another study, IL6 was evaluated among several serum markers in NAFLD patients, and IL6 circulating levels were significantly increased in patients with NAFLD as compared to healthy controls, but not in NASH compared to SS.Citation188
Weight loss resulted in a dramatic decrease of IL6 subcutaneous and hepatic expression with a subsequent reduction in expression of the hepatic suppressor of cytokine signaling 3 (SOCS3) and improved insulin sensitivity. On the other hand, TNFα expression after weight loss only decreased in adipose tissue, not in hepatic tissue.Citation180 This would suggest that the liver might be a key organ for adipose tissue-derived IL6 and TNFα because continuous TNFα/IL6 exposure affects hepatic IR.Citation189
Visfatin
Visfatin, also termed pre-B cell colony enhancing factor (PBEF) or nicotinamide phosphoribosyltransferase (NAMPT) was first identified in 1994 as a protein secreted by activated lymphocytes, synergizing with IL7 and stem cell factor in early B cell formation.Citation190 Although the first discovery of this molecule suggested primarily a cytokine function, its rediscovery as the key enzyme in generating nicotinamide adenine dinucleotide has considerably widened its biological perspective.Citation191 Its extracellular functions (cytokine-like) are mainly proinflammatory as it potently induces various other proinflammatory cytokines such TNFα and IL6. Its intracellular functions concentrate on regulating the activity of NAD-consuming enzymes such as various sirtuins, thereby also affecting TNFα biosynthesis, cell lifespan, and longevity.
Only a few reports have so far addressed the role of this adipocytokine in human NAFLD. In patients with NAFLD, visfatin shows higher serum concentrations and weight loss is associated with both a decrease in serum levels and a reduction in liver mRNA expression, suggesting that the fatty liver might indeed contribute to an observable increase in serum visfatin levels.Citation176 In the same study, immunohistochemistry staining for visfatin was carried out in 18 paired liver biopsies. The staining showed that visfatin was abundantly expressed in hepatocytes; weight loss decreased this expression dramatically. Another report has demonstrated the correlation of visfatin serum levels with liver histology in NAFLD, and such high circulating levels could predict the presence of portal inflammation in NAFLD patients.Citation192
A protective role of visfatin against hepatocyte inflammatory damage was suggested by Jarrar et al.Citation193 In that study, serum visfatin circulating levels in NAFLD patients were higher than in healthy control individuals, both lean and obese without NAFLD. Furthermore, when NASH occurred, visfatin concentration decreased significantly compared with SS, but was still significantly higher than in obese or lean healthy subjects without NAFLD.
Our findings are in line with data reporting that circulating levels and hepatic expression of visfatin are significantly higher in a group of morbidly obese women compared to lean controls and morbidly obese women with normal liver histology. Moreover, serum visfatin correlated well with IL6 and C-reactive protein.Citation194
All these findings suggest that visfatin is a molecule with an important role in the pathophysiology of NAFLD and indicate that the liver could be a major source for this cytokine.
PPARδ
The PPARs family consists of three members: namely, PPARα, PPARβ/δ, and PPARγ. These receptors act as FA sensors that control many metabolic programs that are essential for systematic energy homeostasis. Today, due to its ubiquitous profile, much less is known about PPARδ than the other two in relation to human obesity and NAFLD.Citation195
Oliver et al showed that IR obese rhesus monkeys normalized fasting glucose and insulin, increased high-density lipoprotein cholesterol and reduced low-density lipoprotein (LDL) cholesterol after treatment with a potent and specific PPARδ agonist, the GW501516.Citation196 Other studies in an animal model of adenovirus-mediated hepatic PPARδ overexpression showed that PPARδ regulates lipogenesis and glucose utilization for glycogen synthesis. These effects could result in hepatic protection from FFA-mediated damage, possibly due to the generation of protective MUFA and the lowering of lipotoxic SFA levels.Citation197
Overweight and obese men subjected to the PPARδ agonists, GW501516 or MBX-8025, exhibited improved insulin sensitivity and decreased fasting plasma TGs, non-esterified FAs, apolipoprotein B-100, and LDL-cholesterol, with diminished liver fat content quantified by magnetic resonance imaging.Citation198,Citation199
However, the final mechanisms underlying PPARδ effects in the liver of NAFLD patients still need further study.
NAFLD and lipotoxicity
The pathogenesis of NAFLD is often interpreted by the “double-hit” hypothesis. The primary insult or the “first hit” is lipid accumulation in the liver,Citation8,Citation200 followed by a “second hit” in which proinflammatory mediators induce inflammation, hepatocellular injury, and fibrosis.Citation201 This paradigm suggested TG accumulation to be the “first hit” that predisposes to further liver damage in the pathogenesis of NASH, but has recently been replaced by a more complex model as emerging evidence points to FAs and their metabolites as the true lipotoxic agents.Citation202 Interestingly, lipid accumulation and altered composition of phospholipids within ER membranes further promotes ER stress and IR in obese mice.Citation203 Cytosolic TGs are therefore now considered to be inert, and in fact, lipid droplet accumulation seems to be hepatoprotective.Citation204 However, TG accumulation and lipid droplet formation go hand in hand with pathophysiological mechanisms in NASH. FAs, as well as acyl-CoA and acetyl-CoA, have been identified as potential causes of lipotoxicity.Citation205 FAs have been found to initiate the extrinsic apoptosis cascade and also to interfere with NR signaling, which might influence the extent of hepatocyte damage and further promote IR and ER stress.Citation206 Accordingly, β-oxidation of LCFA within peroxisomes and ω-oxidation within the ER are upregulated in NASH and contribute to lipotoxicity and ROS formation.Citation142 This might be secondary to inhibition of mitochondrial β-oxidation due to an accumulation of malonyl-CoA and the inhibition of CPT1. In fact, recent studies indicate that activation of mitochondrial FA oxidation protects from steatosis and IR.Citation207
It is known that FAs induce the production of TNFα. Hepatic TNF receptor expression correlates with the severity of NAFLD disease.Citation182 Also, TNF receptor activation increases expression of SREBP1c, which induces hepatic lipogenesis and lipid accumulation.Citation208 TNFα activation is further paralleled by death-receptor expression, which facilitates activation of the extrinsic apoptosis cascade. Apoptosis is indeed the predominant form of hepatocellular injury in NASH. In fact, apoptotic activity within the unhealthy liver correlates with disease severity, and thus, cleaved cytokeratin-18 fragments in the serum of NAFLD could effectively be utilized as surrogate markers for the progression of NAFLD.Citation209 As previously mentioned, FA accumulation also leads to the induction of ER stress and ROS formation, which again promotes hepatic injury.Citation210
On the other hand, other studies indicate that metabolic oxidative stress, autophagy, and inflammation are hallmarks of NASH progression. In this sense, CYP2E1, the principal isoform of the CYP450 enzyme, seems to be critically important in NASH development by promoting oxidative/nitrosative stress, protein modifications, inflammation, and IR.Citation211,Citation212 Moreover, Das et al suggest that purinergic receptor X7 (P2X7), upregulated by CYP2E1, might have a key role in autophagy induced by metabolic oxidative stress in NASH.Citation213
In summary, while hepatic TG accumulation seems to be a benign symptom of hepatic steatosis, FA metabolites contribute to the progression of NAFLD to NASH. IR promotes the recruitment of FFAs from the serum pool as well as intrahepatic FA accumulation, which induces apoptosis and ROS formation. FAs themselves also promote hepatic IR via TNF receptor activation, indicating a vicious cycle of lipid accumulation (). Other mechanisms could also contribute to liver damage. Regarding that, some authors have even suggested a “multiple parallel hits hypothesis” to explain the pathophysiology of NAFLD.Citation41
Cannabinoid receptors (CB1, CB2) in NAFLD
The endocannabinoid (EC) system consists of cannabinoid receptors, endogenous cannabinoid ligands, and their biosynthetic and degradative enzymes, and has recently emerged as a ubiquitous system with key functions in a variety of physiological settings. Over the last decade, the EC system has emerged as a pivotal mediator of acute and chronic liver injury. ECs regulate appetite behavior and are lipid mediators that produce similar effects to those of marijuana by acting on membrane-bound receptors.Citation214 Cannabinoid receptors are localized mainly in the brain, but are also present in minor amounts in the liver and some other peripheral tissues (CB1) and in immune and hematopoietic cells (CB2).Citation215,Citation216
ECs may also regulate peripheral energy metabolism, as demonstrated by their CB1-mediated effect on lipoprotein lipase activity in adipocytesCitation217 and their ability to stimulate lipogenesis in hepatocytes.Citation218,Citation219 Cannabinoids exert their effects through two different cannabinoid receptors: CB1 and CB2. Under physiological conditions, the EC system is silent, since CB1 and CB2 receptors are faintly expressed. In contrast, induction of CB receptors and/or increased levels of ECs are common features of liver injuries of diverse origins.Citation220 Both receptors have been implicated in the development of liver fibrosis secondary to various etiologies. CB1-mediated EC tone is enhanced in experimental diet-induced or genetic models of NAFLD, and is characterized by upregulation of adipose tissue and hepatocyte CB1 receptors, and by increased liver synthesis of anandamide. The pathogenic role of CB1 receptors in NAFLD is supported by the resistance to steatosis of obese mice bearing a global or hepatocyte-specific CB1 deletion, or of rodents administered rimonabant or AM6545, a CB1 antagonist.Citation221–Citation223 Studies with cultured hepatocytes and liver slices further indicate that the steatogenic properties of CB1 arise from altered hepatic lipid metabolism, consisting of a combination of hepatocyte activation of SREBP1c-mediated lipogenesis, reduction of FA oxidation via inhibition of AMP kinase, and decreased release of TG-rich VLDL.Citation221,Citation222,Citation224 In addition, the adipose tissue may largely contribute to the steatogenic process via CB1-induced release of FFAs by adipocytes ().Citation225
Figure 5 Mechanisms of CB1 involved in hepatic lipid accumulation.
Abbreviations: ACC1, acetyl-CoA carboxylase; CB, cannabinoid; CPT1, carnitine palmitoyltransferase-1; FA, fatty acid; FAS, fatty acid synthase; FFA, free fatty acid; LPL, lipoprotein lipase; SREBP1c, sterol regulatory element-binding protein 1c; TG, triglyceride; VLDL, very-low-density lipoprotein.
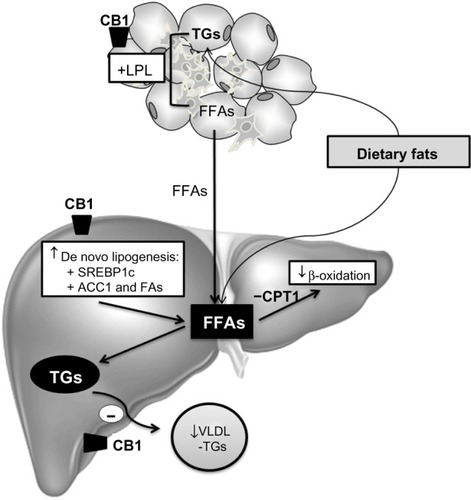
Also, a potential impact of CB1 receptors on the inflammatory response associated with NASH has been suggested by experiments in obese rats showing that rimonabant reduces liver inflammation.Citation222,Citation223 Although the underlying mechanism remains to be delineated, in hepatocytes, CB1 receptors could contribute to the acute phase response, via activation of cAMP responsive element-binding protein (CREBH), a liver-specific transcription factor that upregulates acute phase response genes.Citation226 In addition, fat CB1 receptors reduce the production of adiponectin, an adipokine which reduces hepatic inflammation.Citation222,Citation223
With regard to liver fibrosis, it has been shown that marijuana use may correlate with the progression of fibrosis in patients with hepatitis C.Citation227 However, each receptor seems to have opposing roles in the liver. The CB2 receptor has been shown to be upregulated in the livers of cirrhosis patients and to ameliorate the progression of fibrosis.Citation228 In contrast, CB1 receptor activation has been linked to the progression of fibrosis, and CB1 antagonists have been shown to inhibit the progression of fibrosis.Citation229 Indeed, clinical trials with a CB1 receptor antagonist have shown that antagonism of CB1 can result in weight loss and improved metabolic and cardiac parameters in overweight and obese populations.Citation230 In summary, enhanced CB1 tone promotes liver fibrogenesis and cardiovascular alterations associated with cirrhosis, and contributes to the pathogenesis of NAFLD. On the other hand, upregulated CB2 signaling displays hepatoprotective effects, reducing liver inflammation, and improving liver fibrogenesis. Antagonism of CB1 and agonism of CB2 receptors have been identified as promising therapeutic strategies for the management of liver diseases.
Conclusion
NAFLD is characterized by IR, which leads to the deposition of fat, predominantly TGs, in the liver. The steatotic liver exhibits low-grade liver injury. However, a number of patients develop progressive liver injury with hepatocyte apoptosis, greater oxidative stress, and liver inflammation. The factors that lead to the progression of steatosis to steatohepatitis, are likely to be multiple and complex. We proposed a model of hepatocyte injury in fatty liver: in the susceptible steatotic hepatocyte, circulating FFAs can activate ER stress and apoptosis. While hepatic TG accumulation seems to be a benign symptom of hepatic steatosis, FA metabolites might contribute to the progression of NAFLD to NASH. IR promotes the recruitment of FFAs from the serum pool as well as intrahepatic FA accumulation through altered hepatic lipid metabolism, which finally induces apoptosis and ROS formation. Better understanding of the molecular pathways of liver injury should promote the development of diagnostic and therapeutic interventions aimed at reducing the morbidity and mortality associated with NAFLD.
Disclosure
The authors report no conflicts of interest in this work.
References
- AnguloPObesity and nonalcoholic fatty liver diseaseNutr Rev2007656 Pt 25763
- LazoMClarkJMThe epidemiology of nonalcoholic fatty liver disease: a global perspectiveSemin Liver Dis200828433935018956290
- WilliamsCDStengelJAsikeMIPrevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective studyGastroenterology2011140112413120858492
- NakamutaMKohjimaMMorizonoSEvaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver diseaseInt J Mol Med200516463163516142397
- MachadoMMarques-VidalPCortez-PintoHHepatic histology in obese patients undergoing bariatric surgeryJ Hepatol200645460060616899321
- WieckowskaAPapouchadoBGLiZLopezRZeinNNFeldsteinAEIncreased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitisAm J Gastroenterol200810361372137918510618
- EkstedtMFranzénLEMathiesenULLong-Term Follow-up of Patients with NAFLD and Elevated Liver EnzymesHepatology200644486587317006923
- FarrellGCLarterCZNonalcoholic fatty liver disease: from steatosis to cirrhosisHepatology2006432 Suppl 1S99S11216447287
- DowmanJKTomlinsonJWNewsomePNSystematic review: the diagnosis and staging of non-alcoholic fatty liver disease and non-alcoholic steatohepatitisAliment Pharmacol Ther201133552554021198708
- MarchesiniGBugianesiEForlaniGNonalcoholic fatty liver, steatohepatitis, and the metabolic syndromeHepatology200337491792312668987
- AdamsLAWatersORKnuimanMWElliottRROlynykJKNAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up studyAm J Gastroenterol2009104486186719293782
- AdamsLAFeldsteinAENon-invasive diagnosis of nonalcoholic fatty liver and nonalcoholic steatohepatitisJ Dig Dis2011121101621091933
- YanEDurazoFTongMHongKNonalcoholic fatty liver disease: pathogenesis, identification, progression, and managementNutr Rev2007658 Pt 137638417867371
- SorbiDBoyntonJLindorKDThe ratio of aspartate aminotransferase to alanine aminotransferase: potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver diseaseAm J Gastroenterol19999441018102210201476
- AdamsLAAnguloPLindorKDNonalcoholic fatty liver diseaseCMAJ2005172789990515795412
- FestiDSchiumeriniRMarziLReview article: the diagnosis of non-alcoholic fatty liver disease – availability and accuracy of non-invasive methodsAliment Pharmacol Ther201337439240023278163
- FedericoATrappoliereMLoguercioCTreatment of patients with non-alcoholic fatty liver disease: current views and perspectivesDig Liver Dis2006381178980116750661
- PacificoLCelestreMAnaniaCPaolantonioPChiesaCLaghiAMRI and ultrasound for hepatic fat quantification: relationships to clinical and metabolic characteristics of pediatric nonalcoholic fatty liver diseaseActa Paediatr200796454254717306008
- LewisJRMohantySRNonalcoholic fatty liver disease: a review and updateDig Dis Sci201055356057820101463
- YilmazYUlukayaEDolarEA “biomarker biopsy” for the diagnosis of NASH: promises from CK-18 fragmentsObes Surg2008181115071508 author reply 1509–151018679759
- SchwengerKJAllardJPClinical approaches to non-alcoholic fatty liver diseaseWorld J Gastroenterol20142071712172324587650
- McCarthyEMRinellaMEThe role of diet and nutrient composition in nonalcoholic fatty liver diseaseJ Acad Nutr Diet2012112340140922717200
- PromratKKleinerDENiemeierHMRandomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitisHepatology201051112112919827166
- St GeorgeABaumanAJohnstonAFarrellGCheyTGeorgeJEffect of a lifestyle intervention in patients with abnormal liver enzymes and metabolic risk factorsJ Gastroenterol Hepatol200924339940719067776
- HaukelandJWKonopskiZEggesbøHBMetformin in patients with non-alcoholic fatty liver disease: a randomized, controlled trialScand J Gastroenterol200944785386019811343
- OhMKWinnJPoordadFReview article: diagnosis and treatment of non-alcoholic fatty liver diseaseAliment Pharmacol Ther200828550352218532991
- Van WagnerLBRinellaMEThe role of insulin-sensitizing agents in the treatment of nonalcoholic steatohepatitisTherap Adv Gastroenterol201144249263
- LutchmanGModiAKleinerDEThe effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitisHepatology200746242442917559148
- AithalGPThomasJAKayePVRandomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitisGastroenterology200813541176118418718471
- MaroniLGuastiLCastiglioniLLipid targets during statin treatment in dyslipidemic patients affected by nonalcoholic fatty liver diseaseAm J Med Sci2011342538338721629037
- EkstedtMFranzénLEMathiesenULHolmqvistMBodemarGKechagiasSStatins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up studyJ Hepatol200747113514117400325
- TandraSVuppalanchiRUse of statins in patients with liver diseaseCurr Treat Options Cardiovasc Med200911427227819627660
- ChalasaniNYounossiZLavineJEThe diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological AssociationHepatology20125562005202322488764
- MussoGGambinoRCassaderMRecent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD)Prog Lipid Res200948112618824034
- PettaSMuratoreCCraxìANon-alcoholic fatty liver disease pathogenesis: the present and the futureDig Liver Dis200941961562519223251
- UtzschneiderKMKahnSEReview: the role of insulin resistance in nonalcoholic fatty liver diseaseJ Clin Endocrinol Metab200691124753476116968800
- HotamisligilGSInflammation and metabolic disordersNature2006444712186086717167474
- SavageDBSempleRKRecent insights into fatty liver, metabolic dyslipidaemia and their links to insulin resistanceCurr Opin Lipidol201021432933620581678
- PosticCGirardJContribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered miceJ Clin Invest2008118382983818317565
- LewisGFCarpentierAAdeliKGiaccaADisordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetesEndocr Rev200223220122911943743
- TilgHMoschenAREvolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesisHepatology20105251836184621038418
- KoliwadSKStreeperRSMonettiMDGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammationJ Clin Invest2010120375676720124729
- YamaguchiKYangLMcCallSInhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitisHepatology20074561366137417476695
- AmaroAFabbriniEKarsMDissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemiaGastroenterology2010139114915320303351
- FeldsteinAEWerneburgNWCanbayAFree fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathwayHepatology200440118519415239102
- MaríMCaballeroFColellAMitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitisCell Metab20064318519816950136
- CheungOSanyalAJAbnormalities of lipid metabolism in nonalcoholic fatty liver diseaseSemin Liver Dis200828435135918956291
- MalhiHGoresGJMolecular mechanisms of lipotoxicity in nonalcoholic fatty liver diseaseSemin Liver Dis200828436036918956292
- ArnerPHuman fat cell lipolysis: biochemistry, regulation and clinical roleBest Pract Res Clin Endocrinol Metab200519447148216311212
- DelarueJMagnanCFree fatty acids and insulin resistanceCurr Opin Clin Nutr Metab Care200710214214817285001
- BerkPDRegulatable fatty acid transport mechanisms are central to the pathophysiology of obesity, fatty liver, and metabolic syndromeHepatology20084851362137618972439
- MartinGNemotoMGelmanLThe human fatty acid transport protein-1 (SLC27A1; FATP-1) cDNA and gene: organization, chromosomal localization, and expressionGenomics200066329630410873384
- GeFZhouSHuCLobdellHBerkPDInsulin- and leptin-regulated fatty acid uptake plays a key causal role in hepatic steatosis in mice with intact leptin signaling but not in ob/ob or db/db miceAm J Physiol Gastrointest Liver Physiol20102994G855G86620595619
- ZhouSLStumpDSorrentinoDPotterBJBerkPDAdipocyte differentiation of 3T3-L1 cells involves augmented expression of a 43-kDa plasma membrane fatty acid-binding proteinJ Biol Chem19922672014456144611629231
- ZhouSLStumpDKiangCLIsolaLMBerkPDMitochondrial aspartate aminotransferase expressed on the surface of 3T3-L1 adipocytes mediates saturable fatty acid uptakeProc Soc Exp Biol Med199520832632707878064
- TrigattiBLAndersonRGGerberGEIdentification of caveolin-1 as a fatty acid binding proteinBiochem Biophys Res Commun19992551343910082651
- DoegeHStahlAProtein-mediated fatty acid uptake: novel insights from in vivo modelsPhysiology (Bethesda)20062125926816868315
- DoegeHBaillieRAOrtegonAMTargeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasisGastroenterology200613041245125816618416
- AuingerAValentiLPfeufferMA promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosisHorm Metab Res2010421285484920945272
- FernándezMAAlborCIngelmo-TorresMCaveolin-1 is essential for liver regenerationScience200631357931628163216973879
- MastrodonatoMCalamitaGRossiRAltered distribution of caveolin-1 in early liver steatosisEur J Clin Invest201141664265121250982
- Miquilena-ColinaMELima-CabelloESánchez-CamposSHepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis CGut201160101394140221270117
- InoueMOhtakeTMotomuraWIncreased expression of PPARgamma in high fat diet-induced liver steatosis in miceBiochem Biophys Res Commun2005336121522216125673
- BuquéXMartínezMJCanoAA subset of dysregulated metabolic and survival genes is associated with severity of hepatic steatosis in obese Zucker ratsJ Lipid Res201051350051319783528
- DegracePMoindrotBMohamedIUpregulation of liver VLDL receptor and FAT/CD36 expression in LDLR−/− apoB100/100 mice fed trans-10, cis-12 conjugated linoleic acidJ Lipid Res200647122647265516957181
- ZhouJFebbraioMWadaTHepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosisGastroenterology2008134255656718242221
- GrecoDKotronenAWesterbackaJGene expression in human NAFLDAm J Physiol Gastrointest Liver Physiol20082945G1281G128718388185
- BechmannLPGieselerRKSowaJ-PApoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitisLiver Int201030685085920408954
- FuruhashiMHotamisligilGSFatty acid-binding proteins: role in metabolic diseases and potential as drug targetsNat Rev Drug Discov20087648950318511927
- KarakasSEAlmarioRUKimKSerum fatty acid binding protein 4, free fatty acids, and metabolic risk markersMetabolism20095871002100719394980
- ZimmermanAWVeerkampJHNew insights into the structure and function of fatty acid-binding proteinsCell Mol Life Sci20025971096111612222958
- ChmurzyńskaAThe multigene family of fatty acid-binding proteins (FABPs): function, structure and polymorphismJ Appl Genet2006471394816424607
- KrusinováEPelikánováTFatty acid binding proteins in adipose tissue: a promising link between metabolic syndrome and atherosclerosis?Diabetes Res Clin Pract200882Suppl 2S127S13418977052
- TaskinenMRAdielsMWesterbackaJDual metabolic defects are required to produce hypertriglyceridemia in obese subjectsArterioscler Thromb Vasc Biol20113192144215021778423
- WesterbackaJKolakMKiviluotoTGenes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjectsDiabetes200756112759276517704301
- FalconADoegeHFluittAFATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetaseAm J Physiol Endocrinol Metab20102993E384E39320530735
- DoegeHGrimmDFalconASilencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemiaJ Biol Chem200828332221862219218524776
- NieBParkHMKazantzisMSpecific bile acids inhibit hepatic fatty acid uptake in miceHepatology20125641300131022531947
- OkamuraMInagakiTTanakaTSakaiJRole of histone methylation and demethylation in adipogenesis and obesityOrganogenesis201061243220592862
- SanyalAJChalasaniNKowdleyKVPioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitisN Engl J Med2010362181675168520427778
- FabbriniESullivanSKleinSObesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implicationsHepatology201051267968920041406
- BechmannLPHannivoortRAGerkenGHotamisligilGSTraunerMCanbayAThe interaction of hepatic lipid and glucose metabolism in liver diseasesJ Hepatol201256495296422173168
- KawanoYCohenDEMechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver diseaseJ Gastroenterol201348443444123397118
- FuchsMNon-alcoholic fatty liver disease: the bile acid-activated farnesoid x receptor as an emerging treatment targetJ Lipids2012201293439622187656
- FerréPFoufelleFHepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1cDiabetes Obes Metab201012Suppl 2839221029304
- MitsuyoshiHYasuiKHaranoYAnalysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver diseaseHepatol Res200939436637319054139
- KohjimaMHiguchiNKatoMSREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver diseaseInt J Mol Med200821450751118360697
- BaranowskiMBiological role of liver X receptorsJ Physiol Pharmacol200859Suppl 7315519258656
- FauldsMHZhaoCDahlman-WrightKMolecular biology and functional genomics of liver X receptors (LXR) in relationship to metabolic diseasesCurr Opin Pharmacol201010669269720829110
- LiangGYangJHortonJDHammerREGoldsteinJLBrownMSDiminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1cJ Biol Chem2002277119520952811782483
- PeetDJTurleySDMaWCholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alphaCell19989356937049630215
- RepaJJLiangGOuJRegulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbetaGenes Dev200014222819283011090130
- SchultzJRTuHLukARole of LXRs in control of lipogenesisGenes Dev200014222831283811090131
- HiguchiNKatoMShundoYLiver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver diseaseHepatol Res200838111122112918684130
- Lima-CabelloEGarcia-MediavillaMVMiquilena-ColinaMEEnhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis CClin Sci (Lond)2011120623925020929443
- HortonJDGoldsteinJLBrownMSCritical review SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liverJ Clin Invest200210991125113111994399
- ShimomuraIShimanoHHortonJDGoldsteinJLBrownMSDifferential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cellsJ Clin Invest19979958388459062340
- ShimomuraIBashmakovYIkemotoSHortonJDBrownMSGoldsteinJLInsulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetesProc Natl Acad Sci U S A19999624136561366110570128
- ForetzMPacotCDugailIADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucoseMol Cell Biol19991953760376810207099
- Azzout-MarnicheDBécardDGuichardCInsulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytesBiochem J2000350Pt 238939310947952
- OsborneTFSterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin actionJ Biol Chem200027542323793238210934219
- ChakravartyKLeahyPBecardDSterol regulatory element-binding protein-1c mimics the negative effect of insulin on phosphoenolpyruvate carboxykinase (GTP) gene transcriptionJ Biol Chem200127637348163482311443121
- ShimanoHSterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genesProg Lipid Res200140643945211591434
- HortonJDBashmakovYShimomuraIShimanoHRegulation of sterol regulatory element binding proteins in livers of fasted and refed miceProc Natl Acad Sci U S A19989511598759929600904
- KimJBSarrafPWrightMNutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1J Clin Invest19981011199421459
- NagayaTTanakaNSuzukiTDown-regulation of SREBP-1c is associated with the development of burned-out NASHJ Hepatol201053472473120655124
- DenechaudPDDentinRGirardJPosticCRole of ChREBP in hepatic steatosis and insulin resistanceFEBS Lett20085821687317716660
- FoufelleFFerréPNew perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1cBiochem J2002366Pt 237739112061893
- DentinRPégorierJPBenhamedFHepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expressionJ Biol Chem200427919203142032614985368
- IshiiSIizukaKMillerBCUyedaKCarbohydrate response element binding protein directly promotes lipogenic enzyme gene transcriptionProc Natl Acad Sci U S A200410144155971560215496471
- MaLTsatsosNGTowleHCDirect role of ChREBP.Mlx in regulating hepatic glucose-responsive genesJ Biol Chem200528012120191202715664996
- ChaJYRepaJJThe liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXRJ Biol Chem2007282174375117107947
- UlvenSMDalenKTGustafssonJANebbHILXR is crucial in lipid metabolismProstaglandins Leukot Essent Fatty Acids2005731596315913974
- ChenGLiangGOuJGoldsteinJLBrownMSCentral role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liverProc Natl Acad Sci U S A200410131112451125015266058
- JosephSBLaffitteBPatelPHDirect and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptorsJ Biol Chem200227713110191102511790787
- ZhangYYinLHillgartnerFBSREBP-1 integrates the actions of thyroid hormone, insulin, cAMP, and medium-chain fatty acids on ACCalpha transcription in hepatocytesJ Lipid Res200344235636812576518
- ChuKMiyazakiMManWCNtambiJMStearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activationMol Cell Biol200626186786679816943421
- MitroNMakPAVargasLThe nuclear receptor LXR is a glucose sensorNature2007445712421922317187055
- ArdenCPetrieJLTudhopeSJElevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasisDiabetes201160123110312022013014
- MaLRobinsonLNTowleHCChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liverJ Biol Chem200628139287212873016885160
- de la IglesiaNMukhtarMSeoaneJGuinovartJJAgiusLThe role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyteJ Biol Chem200027514105971060310744755
- IizukaKBruickRKLiangGHortonJDUyedaKDeficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysisProc Natl Acad Sci U S A2004101197281728615118080
- IizukaKMillerBUyedaKDeficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) miceAm J Physiol Endocrinol Metab20062912E358E36416705063
- DentinRBenhamedFHainaultILiver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob miceDiabetes20065582159217016873678
- IizukaKTakedaJHorikawaYHepatic overexpression of dominant negative Mlx improves metabolic profile in diabetes-prone C57BL/6J miceBiochem Biophys Res Commun2009379249950419121288
- BenhamedFDenechaudPLemoineMThe lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humansJ Clin Invest201212262176219422546860
- BrownMSGoldsteinJLSelective versus total insulin resistance: a pathogenic paradoxCell Metab200872959618249166
- FormanBMGoodeEChenJIdentification of a nuclear receptor that is activated by farnesol metabolitesCell19958156876937774010
- LuTTMakishimaMRepaJJMolecular basis for feedback regulation of bile acid synthesis by nuclear receptorsMol Cell20006350751511030331
- ZhangYKast-WoelbernHREdwardsPANatural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activationJ Biol Chem2003278110411012393883
- DownesMVerdeciaMARoeckerAJA chemical, genetic, and structural analysis of the nuclear bile acid receptor FXRMol Cell20031141079109212718892
- ThomasCPellicciariRPruzanskiMAuwerxJSchoonjansKTargeting bile-acid signalling for metabolic diseasesNat Rev Drug Discov20087867869318670431
- KokTHulzebosCVWoltersHEnterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: efficient intestinal bile salt absorption in the absence of ileal bile acid-binding proteinJ Biol Chem200327843419304193712917447
- SinalCJTohkinMMiyataMWardJMLambertGGonzalezFJTargeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasisCell2000102673174411030617
- WatanabeMHoutenSMWangLBile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1cJ Clin Invest2004113101408141815146238
- MaKSahaPKChanLMooreDDFarnesoid X receptor is essential for normal glucose homeostasisJ Clin Invest200611641102110916557297
- ZhangYLeeFYBarreraGActivation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic miceProc Natl Acad Sci U S A200610341006101116410358
- YangZXShenWSunHEffects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver diseaseHepatol Int20104474174821286345
- TeodoroJSRoloAPPalmeiraCMHepatic FXR: key regulator of whole-body energy metabolismTrends Endocrinol Metab2011221145846621862343
- ModicaSGadaletaRMMoschettaADeciphering the nuclear bile acid receptor FXR paradigmNucl Recept Signal20108e00521383957
- DornCRienerMOKirovskiGExpression of fatty acid synthase in nonalcoholic fatty liver diseaseInt J Clin Exp Pathol20103550551420606731
- KohjimaMEnjojiMHiguchiNRe-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver diseaseInt J Mol Med200720335135817671740
- MorganKUyuniANandgiriGAltered expression of transcription factors and genes regulating lipogenesis in liver and adipose tissue of mice with high fat diet-induced obesity and nonalcoholic fatty liver diseaseEur J Gastroenterol Hepatol200820984385418794597
- ZhangWPatilSChauhanBFoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expressionJ Biol Chem200628115101051011716492665
- QuSAltomonteJPerdomoGAberrant Forkhead box O1 function is associated with impaired hepatic metabolismEndocrinology2006147125641565216997836
- MatsumotoMHanSKitamuraTAcciliDDual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolismJ Clin Invest200611692464247216906224
- ValentiLRamettaRDongiovanniPIncreased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitisDiabetes20085751355136218316359
- SamuelVTChoiCSPhillipsTGTargeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin actionDiabetes20065572042205016804074
- NakaeJBiggsWHKitamuraTRegulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1Nat Genet200232224525312219087
- MandardSMüllerMKerstenSPeroxisome proliferator-activated receptor alpha target genesCell Mol Life Sci200461439341614999402
- KerstenSSeydouxJPetersJMGonzalezFJDesvergneBWahliWPeroxisome proliferator-activated receptor alpha mediates the adaptive response to fastingJ Clin Invest1999103111489149810359558
- HaranoYYasuiKToyamaTFenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liverLiver Int200626561362016762007
- IpEFarrellGCRobertsonGHallPKirschRLeclercqICentral role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in miceHepatology200338112313212829994
- ChouCJHaluzikMGregoryCWY14,643, a peroxisome proliferator-activated receptor alpha (PPARalpha) agonist, improves hepatic and muscle steatosis and reverses insulin resistance in lipoatrophic A-ZIP/F-1 miceJ Biol Chem200227727244842448911994294
- HashimotoTCookWSQiCYeldandiAVReddyJKRaoMSDefect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fastingJ Biol Chem200027537289182892810844002
- ReddyJKHashimotoTPeroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic systemAnnu Rev Nutr20012119323011375435
- KashireddyPVRaoMSLack of peroxisome proliferator-activated receptor alpha in mice enhances methionine and choline deficient diet-induced steatohepatitisHepatol Res200430210411015519275
- RaoMSPapreddyKMusunuriSOkonkwoAPrevention/reversal of choline deficiency-induced steatohepatitis by a peroxisome proliferator-activated receptor alpha ligand in ratsIn Vivo200216214515212073774
- IpEFarrellGHallPRobertsonGLeclercqIAdministration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in miceHepatology20043951286129615122757
- TailleuxAWoutersKStaelsBRoles of PPARs in NAFLD: potential therapeutic targetsTriglyceride Metab Dis201218215809818
- Vanden BergheWVermeulenLDelerivePDe BosscherKStaelsBHaegemanGA paradigm for gene regulation: inflammation, NF-kappaB and PPARAdv Exp Med Biol200354418119614713228
- DelerivePDe BosscherKBesnardSPeroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappa B and AP-1J Biol Chem199927445320483205410542237
- DelerivePGervoisPFruchartJCStaelsBInduction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activatorsJ Biol Chem200027547367033670710980195
- GervoisPVu-DacNKleemannRNegative regulation of human fibrinogen gene expression by peroxisome proliferator-activated receptor alpha agonists via inhibition of CCAAT box/enhancer-binding protein betaJ Biol Chem200127636334713347711418615
- GervoisPKleemannRPilonAGlobal suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrateJ Biol Chem200427916161541616014764586
- HuangYYGusdonAMQuSNonalcoholic fatty liver disease: molecular pathways and therapeutic strategiesLipids Health Dis20131217124209497
- TilgHMoschenARInflammatory mechanisms in the regulation of insulin resistanceMol Med2008143–422223118235842
- TilgHAdipocytokines in nonalcoholic fatty liver disease: key players regulating steatosis, inflammation and fibrosisCurr Pharm Des201016171893189520370678
- MaedaNTakahashiMFunahashiTPPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived proteinDiabetes20015092094209911522676
- WolfAMWolfDRumpoldHEnrichBTilgHAdiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytesBiochem Biophys Res Commun2004323263063515369797
- TilgHMoschenARAdipocytokines: mediators linking adipose tissue, inflammation and immunityNat Rev Immunol200661077278316998510
- XuAWangYKeshawHXuLYLamKSCooperGJThe fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in miceJ Clin Invest200311219110012840063
- HuiJMHodgeAFarrellGCKenchJGKriketosAGeorgeJBeyond insulin resistance in NASH: TNF-alpha or adiponectin?Hepatology2004401465415239085
- BaranovaAGowderSJSchlauchKGene expression of leptin, resistin, and adiponectin in the white adipose tissue of obese patients with non-alcoholic fatty liver disease and insulin resistanceObes Surg20061691118112516989692
- KaserSMoschenACayonAAdiponectin and its receptors in non-alcoholic steatohepatitisGut200554111712115591515
- MoschenARMolnarCWolfAMEffects of weight loss induced by bariatric surgery on hepatic adipocytokine expressionJ Hepatol200951476577719664840
- MarraFBertolaniCAdipokines in liver diseasesHepatology200950395796919585655
- HotamisligilGSShargillNSSpiegelmanBMAdipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistanceScience1993259509187917678183
- KernPASaghizadehMOngJMBoschRJDeemRSimsoloRBThe expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipaseJ Clin Invest1995955211121197738178
- MoschenARMolnarCGeigerSAnti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expressionGut20105991259126420660075
- LangCHDobrescuCBagbyGJTumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose outputEndocrinology1992130143521727716
- CrespoJCayónAFernández-GilPGene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patientsHepatology20013461158116311732005
- LesmanaCRAHasanIBudihusodoUDiagnostic value of a group of biochemical markers of liver fibrosis in patients with non-alcoholic steatohepatitisJ Dig Dis200910320120619659788
- ValentiLFracanzaniALDongiovanniPTumor necrosis factor alpha promoter polymorphisms and insulin resistance in nonalcoholic fatty liver diseaseGastroenterology2002122227428011832442
- ZhouYJLiYYNieYQInfluence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese peopleJ Gastroenterol Hepatol201025477277720492333
- GabayCKushnerIAcute-phase proteins and other systemic responses to inflammationN Engl J Med199934064484549971870
- CressmanDEGreenbaumLEDeAngelisRALiver failure and defective hepatocyte regeneration in interleukin-6-deficient miceScience19962745291137913838910279
- HaukelandJWDamåsJKKonopskiZSystemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2J Hepatol20064461167117416618517
- SennJJKloverPJNowakIASuppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytesJ Biol Chem200327816137401374612560330
- SamalBSunYStearnsGXieCSuggsSMcNieceICloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factorMol Cell Biol1994142143114378289818
- MoschenARGernerRRTilgHPre-B Cell colony enhancing factor/NAMPT/visfatin in inflammation and obesity-related disordersCurr Pharm Des201016171913192020370672
- AllerRde LuisDAIzaolaOInfluence of visfatin on histopathological changes of non-alcoholic fatty liver diseaseDig Dis Sci20095481772177719005759
- JarrarMHBaranovaACollantesRAdipokines and cytokines in non-alcoholic fatty liver diseaseAliment Pharmacol Ther200827541242118081738
- AuguetTTerraXPorrasJAPlasma visfatin levels and gene expression in morbidly obese women with associated fatty liver diseaseClin Biochem201346320220823174488
- VidelaLAPettinelliPMisregulation of PPAR functioning and its pathogenic consequences associated with nonalcoholic fatty liver disease in human obesityPPAR Res2012201210743423304111
- OliverWRJrShenkJLSnaithMRA selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transportProc Natl Acad Sci U S A20019895306531111309497
- LiuSHatanoBZhaoMRole of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulationJ Biol Chem201128621237124721059653
- RisérusUSprecherDJohnsonTActivation of peroxisome proliferator-activated receptor (PPAR)delta Promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese menDiabetes200857233233918024853
- OoiEMMWattsGFSprecherDLChanDCBarrettPHRMechanism of action of a peroxisome proliferator-activated receptor (PPAR)-delta agonist on lipoprotein metabolism in dyslipidemic subjects with central obesityJ Clin Endocrinol Metab20119610E1568E157621816786
- HaiderDGSchindlerKSchallerGPragerGWolztMLudvikBIncreased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric bandingJ Clin Endocrinol Metab20069141578158116449335
- TackeFLueddeTTrautweinCInflammatory pathways in liver homeostasis and liver injuryClin Rev Allergy Immunol200936141218600481
- Neuschwander-TetriBAHepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolitesHepatology201052277478820683968
- FuSYangLLiPAberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesityNature2011473734852853121532591
- RicchiMOdoardiMRCarulliLDifferential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytesJ Gastroenterol Hepatol200924583084019207680
- HanMSParkSYShinzawaKLysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytesJ Lipid Res2008491849717951222
- NolanCJLarterCZLipotoxicity: why do saturated fatty acids cause and monounsaturates protect against it?J Gastroenterol Hepatol200924570370619646010
- Orellana-GavaldaJMHerreroLMalandrinoMIMolecular therapy for obesity and diabetes based on a long-term increase in hepatic fatty-acid oxidationHepatology201153382183221319201
- EndoMMasakiTSeikeMYoshimatsuHTNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c)Exp Biol Med (Maywood)2007232561462117463157
- FeldsteinAEWieckowskaALopezARLiuYCZeinNNMcCulloughAJCytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation studyHepatology20095041072107819585618
- FeldsteinAENovel insights into the pathophysiology of nonalcoholic fatty liver diseaseSemin Liver Dis201030439140120960378
- AbdelmegeedMABanerjeeAYooSHCritical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitisJ Hepatol201257486086622668639
- SethRKDasSKumarACYP2E1-dependent and leptin-mediated hepatic CD57 expression on CD8+ T cells aid progression of environment-linked nonalcoholic steatohepatitisToxicol Appl Pharmacol20142741425424211274
- DasSSethRKKumarAPurinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitisAm J Physiol Gastrointest Liver Physiol201330512G950G96324157968
- Di MarzoVPiscitelliFMechoulamRCannabinoids and endocannabinoids in metabolic disorders with focus on diabetesHandb Exp Pharmacol20112037510421484568
- PacherPMechoulamRIs lipid signaling through cannabinoid 2 receptors part of a protective system?Prog Lipid Res201150219321121295074
- PacherPBátkaiSKunosGThe endocannabinoid system as an emerging target of pharmacotherapyPharmacol Rev200658338946216968947
- CotaDMarsicanoGTschapMThe endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesisJ Clin Invest2003112342343112897210
- Osei-HyiamanDDePetrilloMPacherPEndocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesityJ Clin Invest200511551298130515864349
- JeongWOsei-HyiamanDParkOParacrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liverCell Metab20087322723518316028
- MallatATeixeira-ClercFDeveauxVManinSLotersztajnSThe endocannabinoid system as a key mediator during liver diseases: new insights and therapeutic openingsBr J Pharmacol201116371432144021457226
- Osei-HyiamanDLiuJZhouLHepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in miceJ Clin Invest200811893160316918677409
- TamJVemuriVKLiuJPeripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesityJ Clin Invest201012082953296620664173
- Gary-BoboMElachouriGGallasJFRimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa ratsHepatology200746112212917526015
- JourdanTDemizieuxLGrestiJAntagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: evidence from cultured explantsHepatology201255379079921987372
- JourdanTDjaoutiLDemizieuxLGrestiJVergèsBDegracePCB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese miceDiabetes201059492693420110567
- ChandaDKimDKLiTCannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytesJ Biol Chem201128632279712797921693703
- HézodeCRoudot-ThoravalFNguyenSDaily cannabis smoking as a risk factor for progression of fibrosis in chronic hepatitis CHepatology2005421637115892090
- Muñoz-LuqueJRosJFernández-VaroGRegression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic ratsJ Pharmacol Exp Ther2008324247548318029545
- Teixeira-ClercFJulienBGrenardPCB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosisNat Med200612667167616715087
- Van GaalLPi-SunyerXDesprésJPMcCarthyCScheenAEfficacy and safety of rimonabant for improvement of multiple cardiometabolic risk factors in overweight/obese patients: pooled 1-year data from the Rimonabant in Obesity (RIO) programDiabetes Care200831Suppl 2S229S24018227491