
Abstract
Genomic deoxyribonucleic acid (DNA) is under constant threat from endogenous and exogenous DNA damaging agents. Mammalian cells have evolved highly conserved DNA repair machinery to process DNA damage and maintain genomic integrity. Impaired DNA repair is a major driver for carcinogenesis and could promote aggressive cancer biology. Interestingly, in established tumors, DNA repair activity is required to counteract oxidative DNA damage that is prevalent in the tumor microenvironment. Emerging clinical data provide compelling evidence that overexpression of DNA repair factors may have prognostic and predictive significance in patients. More recently, DNA repair inhibition has emerged as a promising target for anticancer therapy. Synthetic lethality exploits intergene relationships where the loss of function of either of two related genes is nonlethal, but loss of both causes cell death. Exploiting this approach by targeting DNA repair has emerged as a promising strategy for personalized cancer therapy. In the current review, we focus on recent advances with a particular focus on synthetic lethality targeting in cancer.
Keywords:
Introduction
Genomic deoxyribonucleic acid (DNA) is at risk of damage from endogenous metabolic byproducts, spontaneous base modifications, and exogenous sources such as ultraviolet (UV) light, ionizing radiation, and chemical agents. Unrepaired DNA damage is a major source of potentially mutagenic lesions that drive carcinogenesis. To promote genomic stability, mammalian cells have evolved highly conserved DNA damage sensor mechanisms that can initiate: 1) induction of apoptosis to eliminate heavily damaged cells; 2) transcriptional response, which causes changes in the transcriptional profile that may promote cell survival; 3) DNA damage tolerance; 4) activation of DNA damage checkpoints and modulation of cell-cycle progression to allow time for DNA repair; and/or 5) initiation of DNA repair to restore genomic stability.
DNA repair pathways are so essential that germline mutations within DNA repair genes are associated with cancer predisposition syndromes such as hereditary non-polyposis carcinoma coli (HNPCC) or breast cancer susceptibility protein (BRCA)-deficient breast and ovarian cancer syndromes.Citation1,Citation2 In addition, polymorphic variants that confer suboptimal DNA repair capacity could also influence cancer susceptibility and prognosis.Citation3,Citation4 Furthermore, the anticancer activity of chemotherapy (such as alkylating agents and platinum compounds) and radiotherapy is to a large extent directly related to their ability to induce DNA damage. The DNA repair capacity of cancer cells to recognize and repair chemo/radiotherapy-induced damage is therefore also an important mechanism for therapeutic resistance that negatively impacts upon clinical outcomes.Citation5 Pharmacological inhibition of DNA repair may increase cytotoxicity of anticancer agents, reverse treatment resistance and improve therapeutic efficacy.Citation6 Recent evidence indicates that inhibitors of DNA repair also offer the opportunity to target genetic differences that exist between normal and tumor tissue.Citation7,Citation8
In the current review, we provide an overview of major DNA repair pathways in mammalian cells. We will then focus on the emerging DNA repair drug targets for personalized cancer therapy.
DNA repair pathways
DNA repair pathways operate in mammalian cells to maintain genomic integrity (). Loss of efficiency of one or more DNA repair mechanisms, whether by germline inheritance or sporadic mutation, accelerates the rate of accumulation of additional mutations by 100–1,000 times, with selective pressure favoring those mutations that drive carcinogenesis – the “mutator phenotype”.Citation9,Citation10
Figure 1 Types of DNA damage and DNA repair pathways.
Abbreviation: DNA, deoxyribonucleic acid.
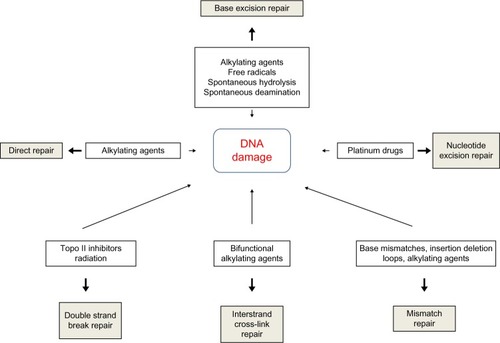
Direct repair
A number of mechanisms exist to directly reverse certain DNA-damage lesions in single-step processes. Direct reversal of the oxidative lesion O6-methylguanine is carried out by the suicide enzyme methylguanine methyltransferase (MGMT) via an active site Cys145 that acts as a methyl recipient, followed by rapid ubiquitin-induced degradation. MGMT expression is one of several factors governing response to alkylating chemotherapy agents.Citation11,Citation12 The 2-oxoglutarate/iron-dependent dioxygenases, alkylated DNA repair protein alkB homolog 2 (ABH2) and alkylated DNA repair protein alkB homolog 3 (ABH3) also repair various alkylation adducts, including 1-methyladenine and 1-ethyladenine, by oxidative dealkylation. ABH2 preferentially acts on double-stranded DNA, possibly in the vicinity of replication forks, whereas ABH3 is involved in single-stranded DNA and ribonucleic acid (RNA) repair.Citation11 ABH2 and ABH3 knockout mice are viable with no cancer phenotype, although ABH2-deficient mice do spontaneously accumulate 1-methyladenine adducts and are hypersensitive to exogenous alkylating agents.Citation13 UV-induced damage such as cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6–4) pyrimidone photoproducts (6–4PPs) are repaired via photolyases: 50–55 kDa flavoproteins that contain chromophoric groups (flavin adenine dinucleotides) that become activated when illuminated with visible or near-UV light, allowing transfer of an electron to the lesion to destabilize the interpyrimidine bonds.Citation14
Base excision repair
Base excision repair (BER) is responsible for detection and repair of damage caused by a number of mechanisms, including alkylation, oxidation, ring saturation, single-strand breaks (SSBs), and base deamination. Although complex, with at least two subpathways (short patch and long patch), BER generally proceeds via: 1) recognition and removal of a damaged base by a DNA glycosylase to form an apurinic/apyrimidinic (AP) site intermediate, 2) cleavage of the phosphodiester backbone 5′ to the AP site by AP endonuclease 1 (APE1), 3) removal of the 5′ sugar fragment, 4) incorporation of the correct base by a DNA polymerase, and 5) sealing of the strand break by a DNA ligase ().Citation14–Citation21 Given the wide range of substrate lesions and potential mutagenic sequelae of failed repair, several BER gene mutations have been linked to human disease, including autosomal recessive familial adenomatous polyposis (MYH mutation), primary immunodeficiency disorders (uracil DNA glycosylase mutation), and neurological disorders (mutations in auxiliary genes such as aprataxin, tyrosyl-DNA phosphodiesterase 1, or polynucleotide kinase 3′-phosphatase). Furthermore, large numbers of single-nucleotide polymorphisms in BER genes have been identified, with variable effect on repair capacity and pathological consequences (reviewed in Wilson et al).Citation22
SSB repair
SSB repair (SSBR) is most accurately considered a BER-related pathway, given the similarity of substrates and shared protein members. SSBR repairs single-strand discontinuities arising from a variety of sources, including reactive oxygen species (ROS), base deamination, and BER intermediates. It also repairs breaks introduced by DNA topoisomerase 1 (topo I) activity, which transiently introduces a DNA nick to relax DNA during transcription and replication, but which can fail to reseal the nick if in close proximity to polymerases or other DNA lesions.Citation23,Citation24 SSBR requires effective surveillance and damage detection, for which PARP1 (poly[ADP-ribose] polymerase 1) is believed to play an essential role. On detecting an SSB, PARP1 rapidly becomes bound and poly(ADP-ribosyl)ated, protecting the nick ends from undesirable recombination and allowing the recruitment of the molecular scaffold protein X-ray repair cross-complementing protein 1 (XRCC1) for ongoing repair. As with BER, end processing follows damage recognition and may be undertaken by a large range of proteins (depending upon the termini damage present), each of which requires interaction with XRCC1 for efficient activity. ROS-related damage often results in 3′-phosphate and 3′-phosphoglycolate modifications, which are processed by polynucleotide kinase (PNK) 3′-phosphatase (PNKP) and APE1 respectively. Topo I-associated SSBs require processing by TDP1 (tyrosyl-DNA phosphodiesterase 1), whereas 5′-adenosine monophosphate-SSBs resulting from abortive DNA ligase activity at existing SSBs are processed by aprataxin. Repair can then proceed via short- or long-patch gap filling and end ligation as in the classical BER pathway.Citation25 SSBR may also play a role in replication-associated damage repair.Citation25,Citation26 When the replication machinery encounters an unrepaired SSB, fork collapse occurs, with the creation of a one-ended double-strand break (DSB) on one chromatid, and an SSB on the other. The DSB is processed by components of the homologous recombination (HR) pathway to allow RAD51-mediated template switching and reformation of the replication fork. Without repair, the associated SSB would be converted to a further DSB on replication fork restart, and hence would represent an irrevocably unrepairable lesion. SSBR end-processing and long-patch BER are probably involved in replication-coupled SSBR, as highlighted by the transcriptional activation of the critical SSBR enzyme XRCC1 by replication-associated transcription factors, such as forkhead box protein M1 (FOX M1) and E2F-1.Citation27,Citation28
Nucleotide excision repair
Nucleotide excision repair (NER) recognizes and repairs base lesions associated with distortion of the DNA helical structure, including UV-induced photoproducts not eliminated by direct repair, and an array of bulky adducts induced by various exogenous chemical agents. Two subpathways of NER exist: global genome NER (GG-NER) and transcription-coupled NER (TC-NER). TC-NER removes lesions from the transcribed DNA strand of transcriptionally active genes when encountered by RNA polymerase II, restoring transcriptional activity and preventing apoptosis. GG-NER performs this process with poor efficiency, instead removing lesions on non-transcribed strands and transcriptionally inert genes to avoid replication fork stalling and chromosomal breakages.Citation29 In GG-NER, damage recognition is sensed by various proteins, including the xeroderma pigmentosum (XP), complementation group C (XPC)-RAD23B complex (helix distortions), UV-damaged DNA-binding protein 1 (DDB1), and UV-damaged DNA-binding protein 2 (DDB2) (UV damage), and XPA (unknown substrate).Citation30 In TC-NER, recognition is mediated by stalling of RNA polymerase II at a damaged site. Recognition factor binding in both pathways is associated with localized distortion to allow repair factor access to the damaged site. Transcription factor IIH (TFIIH), a nine-subunit complex including the DNA helicases XP complementation group B (XPB) and XP complementation group D (XPD), is recruited to unwind the DNA local to the damaged site. Dual incision around the lesion is performed by structure-specific endonucleases XP complementation group G (XPG) (3′ incision) and the excision repair cross-complementing group 1 (ERCC1)-XP complementation group F (XPF) complex (5′ incision), resulting in cleavage of a 24–29 nucleotide fragment. In common with BER and mismatch repair (MMR), proliferating cell nuclear antigen (PCNA) is then recruited to coordinate DNA polymerase repair synthesis and DNA ligase nick joining.Citation29 Given the critical role NER plays in repairing UV-induced damage, it is unsurprising that mutation within NER genes can lead to UV hypersensitivity. XP is an autosomal recessive syndrome that manifests as photosensitivity, neurological abnormalities, and predisposition to skin and other cancers. XP is characterized by marked clinical and genetic heterogeneity, with causative mutations falling into one of seven complementation groups that span many NER factors. Different mutations in the same NER genes can give rise to alternative phenotypes that do not include cancer predisposition, namely Cockayne syndrome (variable UV sensitivity, premature aging, and physical and mental retardation) and trichothiodystrophy (TTD; variable UV-sensitivity, premature aging, ichthyosis, brittle hair, and short stature).Citation31 This heterogeneity may result from the bifunctionality of NER factors. For example, the XPD complementation group is associated with several disease-specific mutations that may cause XP, combined Cockayne syndrome and XP, or TTD. When the causative mutation is associated with XP, the NER function of TFIIH is deficient, correlating with a phenotype of severe UV-sensitivity and cancer predisposition. TFIIH also has a role in transcriptional initiation of RNA polymerase II, in which the helicase subunits (XPB and XPD) unwind the DNA at the promoter region to allow transcription complex access. XPD mutations affecting this function may have normal NER (non-UV-sensitive TTD), or defective CPD but intact 6–4PP repair (UV-sensitive TTD), accounting for the reduced malignancy risk.
MMR
MMR recognizes and repairs errors introduced during replication. DNA polymerases possess 3′–5′ exonuclease activity to excise incorrectly paired bases in newly synthesized DNA. Failure of this proofreading process leads to mispair persistence, forming a substrate for MMR. MMR also recognizes and repairs insertion/deletion loops (IDLs), particularly within microsatellite DNA – hence, “microsatellite instability” is recognized as a hallmark of MMR failure.Citation32,Citation33 If microsatellite instability manifests within tumor suppressor genes, it can produce frame-shift mutations that contribute to carcinogenesis – a common feature of certain cancers, including colorectal, endometrial, ovarian, and gastric cancer (reviewed in Li).Citation34 MMR error recognition is initiated by heterodimers of MSH (Escherichia coli MutS homolog) ATPases. MutSα comprises MSH2 and MSH6, and recognizes base mismatches and small 1–2 nucleotide IDLs. MutSβ, comprising MSH2 and MSH3, binds to larger IDLs of up to 16 nucleotides. Loss of MSH2 results in the absence of both MMR subpathways, and hence cancer predisposition in preclinical modelsCitation35 and in the clinical setting (as HNPCC).Citation36 Once bound to the DNA substrate, the MutS heterodimer recruits MutLα, a heterodimer comprised of MutL homolog 1 (MLH1) and PMS2, homologs of the E. coli ATPase MutL. MLH1 is critical to MMR, with deletion or promoter methylation producing a comparable phenotype to MSH2 mutation.Citation37,Citation38 The MutS/MutL complex functions as a sliding clamp, undergoing an ATP-dependent conformational change that results in release from the mismatch site followed by translocation in both directions from the mismatch. Translocation continues until a strand break is encountered, such as the 3′ terminus of the leading strand or the 5′ or 3′ termini of Okazaki fragments (200–1,000 nucleotide lagging strand fragments generated and later ligated during replication). Exonuclease-1 (EXO1) is loaded at the strand break and degrades the strand back towards the mismatch site, allowing repair using the parent strand template by the high fidelity DNA polymerase δ (Polδ). DNA ligase I seals the gap.Citation33,Citation34
Non-homologous end joining
Along with HR repair, non-homologous end-joining is one of the two major pathways that exist for repair of DSBs. Nonhomologous end joining (NHEJ) essentially involves processing of the terminal nucleotides to allow end ligation, in a manner that restores molecular integrity but may not maintain sequence fidelity. Damage recognition in NHEJ is performed by the Ku70/Ku80 heterodimer, which binds to the DSB ends with high affinity, possibly tethering the broken ends together. Ku binding recruits and activates the DNA-dependent protein kinase DNA-PKcs, forming the DNA-PK complex that phosphorylates other repair proteins including XRCC4-like factor (XLF, also known as Cernunnos), Werner syndrome helicase (WRN), DNA ligase IV (LIG4), and XRCC4. Additionally, DNA-PK is able to autophosphorylate, allowing NHEJ regulation. DNA end-processing prepares damaged terminal nucleotides for ligation. Dependent on the nature of the damage, this may require variable combinations of repair factors. Damaged DNA overhangs can be removed by nucleases such as Artemis, while other factors such as WRN, PNK, and aprataxin and PNK-like factor also have roles. Nucleotide incorporation by DNA polymerases λ and μ replaces nucleotides cleaved during end-processing in a non-template dependent manner, and represents a significant source of error in the pathway. Once processed, ends are ligated by the LIG4/XRCC4/XLF complex.Citation39–Citation41
NHEJ is able to resolve a large variety of DNA DSB damage, and is thought to be the major pathway for DSB repair in eukaryotes, occurring throughout the cell cycle. Accurate repair in NHEJ is likely if the DSB possesses fully complementary single-stranded ends. In the absence of complementary ends, repair is dependent on annealing of short microhomologous sequences (four or fewer nucleotides) within the overhanging ends, which leads to the introduction of potentially mutagenic deletions or insertions.Citation42 NHEJ also has a critical role in V(D)J recombination to recombine the variable region in B-cell and T-cell receptors – in this context, low fidelity is beneficial to maximize diversityCitation43
Microhomology-mediated end joining
The observation that NHEJ can proceed in the absence of key factors, including DNA-PKcs and Ku, without significant input from the HR pathway, has led to the theory that at least one backup NHEJ pathway exists.Citation44 It appears that this pathway functions when classical NHEJ fails, either due to enzymatic deficiency or failure to interact at certain DNA lesions. This alternate mechanism, known as microhomology-mediated end joining (MMEJ), is reliant on the annealing of microhomologous regions of 5–25 base pairs. To uncover these microhomologies, binding of the NHEJ heterodimer Ku and the HR factor RAD51 must be inhibited, possibly by members of the PARP family, to allow 5′–3′ nucleolytic resection by the MRN complex (MRE11-RAD50-NBS1). During this process, replication protein A (RPA) binds to the single-stranded DNA ends to prevent self-complementization. Once microhomologous regions are uncovered, the DNA overhangs anneal. End processing proceeds as required, via XRF-ERCC1 flap cleavage and polymerase-mediated gap-filling. Ligation appears to involve DNA ligase I (LIG1) and III (LIG3).Citation42,Citation45 Unlike NHEJ, MMEJ is always associated with sequence loss and is therefore always mutagenic. Indeed, microhomologies can frequently be demonstrated at chromosome breakpoints in human cancer cells.Citation45
Single-strand annealing
Like MMEJ, single-strand annealing also involves 3′ end resection to uncover homologous regions that can anneal directly under RAD52 control. The size of homologous repeat sequences utilized in single-strand annealing (such as ~300 bp Alu repeats) requires more extensive end resection creating a large flap. Flap removal is catalyzed by RAD1/RAD10 nucleases, under SAW1/SLX4 (single-strand annealing weakened 1/structure-specific endonuclease subunit 4) guidance, resulting in a large deletion event. Significant sequence diversity within repeat elements probably suppresses this pathway to a relatively minor role in DSB repair.Citation40,Citation46–Citation60
HR
HR utilizes a homologous DNA sequence as a template for DNA synthesis and gap filling to ensure error-free repair.Citation40,Citation46–Citation60 For this reason, HR is the predominant mechanism for DSB repair during cellular replication. Cell cycle control is exerted by a dependence on cyclin-dependent kinase activity, which is upregulated in S and G2 phases, although high levels in M phase are associated with HR suppression due to concurrent BRCA2 phosphorylation. HR is also suppressed during G1, when use of the homologous chromosome as a template would result in loss of heterozygosity. Pathway choice for DSB repair is probably also guided by the repair substrate. Two-ended DSBs formed by fracture of a duplex molecule can generally be accurately repaired by simple end ligation via the NHEJ pathway, which in mammalian cells is the predominant mechanism of repair of this type of damage.Citation61 Conversely, one-ended DSBs occurring when a replication fork encounters an SSB or distorting base lesion require template-guided repair to prevent inappropriate annealing leading to large-scale rearrangements or insertions/deletions.
In general, HR requires: 1) damage recognition, 2) end resection mediated by the MRN complex, 3) RAD51-dependent homology-directed strand invasion and repair synthesis, 4) dissociation from the template strand, and 5) end ligation. The classical model is synthesis-dependent strand annealing, which occurs at two-ended DSBs. Following damage recognition, 3′ strand resection coordinated by the MRN complex occurs on both fractured strands. MRN interacts with CtIP (also known as RBBP8 [retinoblastoma binding protein 8]) to promote end resection and generate 3′ single-strand DNA overhangs. These are bound by the protective RPA to prevent self-annealing. RAD51 binds the DNA ends, in combination with associated proteins including BRCA2, RAD52, RAD54, RAD54B, and the RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3). The resultant nucleoprotein filament invades the sister chromatid or homologous chromosome to search for homologous regions to form heteroduplex DNA. Polymerase η catalyzes 3′ extension of the invading strand using the sister strand as a template. The non-template DNA strand is displaced into a D-loop, with a Holliday junction forming at the crossover between the hetero- and homoduplex DNA. This junction can slide along the DNA in either direction (“branch migration”), facilitated by numerous proteins (WRN, BLM, p53, RAD54, BLAP75, MSH2/MSH6) via mechanisms that are yet to be elucidated. Branch migration results in strand dissociation upon reaching the terminus of the invading strand. Once released, the newly synthesized strand anneals beyond the original breakpoint, where it is bound by RPA and RAD52 to coordinate recruitment of factors involved in end processing (eg, XPF/ERCC1 for flap removal), gap filling (by DNA polymerases) and end ligation (by DNA ligases). The repaired strand can then be used as a template for DNA synthesis on the non-invading strand. As a result, sequence information is copied from the template region into the breakpoint (“gene conversion”), potentially resulting in loss of heterozygosity. An alternative model theorizes the formation of a double Holliday junction through simultaneous strand invasion by both DSB 3′ ends, possibly as a mechanism for DSB repair during meiosis. Strand dissociation in this model requires cleavage at the Holliday junction. Depending on the orientation of cleavage, this might result in a crossover event, which could account for the large-scale sequence exchanges that can be demonstrated in meiotic cells.
It is now thought that the primary repair substrates for HR are one-ended DSBs formed by replication fork collapse at the site of an unrepaired SSB or base lesion. Repair of such lesions occurs by the break-induced replication pathway, which initially proceeds in a similar manner to two-ended DSB repair, with 3′ resection creating an overhang which invades the sister chromatid and anneals to a homologous region in a RAD51-mediated mechanism. Cleavage of the single Holliday junction restores the replication fork, allowing replication to continue. Dependent on the orientation of Holliday junction cleavage, this mechanism can result in the leading strand template becoming ligated to the newly synthesized lagging strand, resulting in a sister chromatid exchange.
Evidence to support replication fork collapse as the primary substrate for HR is based upon comparison of the recombination products formed following repair of induced SSBs and spontaneous recombination events. Restriction endonuclease-induced two-ended DSBs result in short tract gene conversion events, consistent with the synthesis-dependent strand annealing model of HR described above. In contrast, camptothecin exposure induces one-ended DSBs by stabilizing DNA-topo-I interactions to prevent re-ligation of topo I-induced SSBs, thus leading to replication fork collapse. Repair of camptothecin-induced damage results in sister chromatid exchanges and long tract gene conversions. Similarly, impaired repair of SSBs in XRCC1- or PARP1 -deficient cells, or following PARP inhibitor exposure, increases one-ended DSB formation, associated with increased formation of γH2AX and RAD51 foci – markers of HR activity.Citation8,Citation62 Spontaneous recombination outcomes are more similar in spectrum to failed SSBR or camptothecin-related repair products than to those formed during the repair of endonuclease-induced two-ended DSBs, suggesting that replication fork collapse forms the primary repair substrate for HR.Citation62
Crosslink repair
A number of DNA repair mechanisms play a role in the repair of interstrand crosslinks (ICLs), a highly toxic form of damage that can stall and collapse replication forks, potentially leading to DNA rearrangement, mutation, or cell death. ICLs are cytotoxic at densities as low as 40 per cell, because they cause DNA distortion and prevent strand dissociation, impacting upon DNA synthesis and replication.Citation63
Fanconi anemia (FA) is an autosomal recessive condition associated with predisposition to acute myelogenous leukemia and other malignancies, progressive bone marrow failure, short stature, and developmental delay. Fourteen complementation groups have been identified, with evidence suggesting that an FA core complex containing Fanconi anemia, complementation group A (FANCA), FANCB, FANCC, FANCE, FANCF, FANCG, and FANCL localizes to DNA damage and activates FANCD2, which in turn co-localizes with BRCA1. Recent studies also implicate XPF mutations in FA.Citation64,Citation65 Given that FA is associated with hypersensitivity to crosslinking agents, it is believed that the genetic basis may be a defect in ICL repair, although much of the pathway remains to be elucidated. The covalent link of the ICL causes localized DNA distortion and prevents replication-mediated DNA unwinding, leading to replication fork stalling. This is recognized by FANCM and associated proteins, which recruits the FA core complex and other repair proteins, including FANCD2-FANCI. The FA core complex possesses ubiquitin ligase activity, which monoubiquitinates FANCD2-FANCI, allowing interaction with Fanconi-associated nuclease 1 (FAN1) and DNA polymerase ν (Polν, POLN). FAN1 has 5′–3′ exonuclease and 5′-flap endonuclease activity, causing DNA cleavage (known as “unhooking”) alongside the ICL, converting the stalled replication fork into a one-ended DSB on the sister chromatid. A number of other nucleases may mediate DNA cleavage 3′ to the ICL, including MUS81-EME1, XPF-ERCC1, and SLX1-SLX4, thus excising the damaged region. PCNA is recruited, coordinating a switch to translesion synthesis (TLS) involving REV1 and DNA polymerase ζ to extend the nascent strand beyond the ICL site.Citation66 Subsequently, the 3′ strand of the sister chromatid DSB is reintegrated into the homologous duplex, forming a double Holliday junction, and replication is restarted via HR in a RAD51-dependent manner.Citation67–Citation70
DNA damage tolerance mechanisms
Unrepaired lesions (particularly bulky adducts, intercalations, crosslinks, and helix distortions) can block progression of the replication fork. DNA damage tolerance mechanisms allow the replication machinery to bypass these lesions prior to repair. This allows replication to proceed without affecting the viability of dividing cells, but can increase the risk of propagating mutations to the daughter population.Citation71
The DNA polymerase-mediated TLS pathway may proceed by one of two models. In polymerase-switching TLS, fork progression is stalled when the replicative helicase encounters a DNA lesion. This triggers the recruitment of specialized TLS polymerases that insert one or more nucleotides opposite the damaged base before the replicative polymerase is “switched” back into the replication machinery. The alternative “gap-filling” TLS model involves reinitiation of replication downstream from the damage lesion, resulting in a single-strand gap that is filled by one of the TLS polymerases. Polymerases that have been implicated in TLS include Polν, Polθ (A family polymerases), Polζ (B family polymerase), Polβ, Poλ, Pohμ, and terminal deoxynucleotidyl transferase (X family polymerases), Polη, Polκ, Polι, and Rev1 (Y family polymerases). Polymerase choice may be damage-specific: for example, Polη) may bypass UV-induced CPDs, whereas Rev1 may function in AP site bypass.Citation72 TLS fidelity is polymerase-specific, highly variable, and may be an important source of genomic instability and susceptibility to cancer and other diseases.Citation73
The HR pathway may also play a role in bypassing unrepaired lesions during replication, via a subpathway known as template switching. In this mechanism, the stalled daughter strand invades the sister chromatid in an HR-mediated mechanism to utilize the complementary parent strand as a template for synthesis, creating a Holliday junction that is resolved prior to resumption of replication. An alternative HR-related model is that of fork regression. The presence of a single-strand base lesion blocks replication on one parent strand, whilst replication persists on the complementary strand beyond the point of damage. Regression of the replication fork allows transient annealing of the daughter strands into a Holliday intermediate (“chicken foot” structure), providing an alternative template for synthesis across the damaged region.Citation74,Citation75
Global DNA damage response
The cellular DNA damage response involves activation of cell cycle checkpoints to induce a cell cycle arrest while repair mechanisms, transcriptional modulation, and/or apoptotic pathways are activated. DNA damage is detected by sensor proteins, which may overlap with specific repair pathway damage sensors. Checkpoint-specific sensors include: ataxia telangiectasia mutated (ATM), which primarily detects DSBs; ataxia telangiectasia and Rad3 related (ATR), which detects UV-induced and other damage; and RAD17-replication factor C (RFC complex) in conjunction with the RAD9-RAD1-HUS1 (9-1-1) complex, which can detect multiple damage types. Damage sensors interact with a wide range of mediator proteins, including BRCA1, MDC1 (mediator of DNA damage checkpoint 1), 53BP1 (p53 binding protein 1), and Claspin, which are required for downstream activation of Chk1 (activated downstream from ATR signaling) and Chk2 (activated downstream from ATM signaling). Chk1 and −2 kinases phosphorylate the phosphotyrosine phosphatases Cdc25A, −B, and −C, leading to their inactivation. As a result, the Cdc phosphatases are unable to dephosphorylate the cyclin-dependent kinases that promote cell cycle transition: Cdk2, which promotes the G1/S transition, and Cdc2 phosphotyrosine, which promotes the G2/M transition.
Three main checkpoints exist: G1/S, intra-S phase, and G2/M. The G1/S checkpoint is activated by damage that prevents initiation of replication, via the ATM-Chk2-Cdc25A or ATR-Chk1-Cdc25A pathways, and is maintained by Chk1 or Chk2-mediated phosphorylation of p53, which leads to p21-mediated inactivation of Cdk2. The intra-S phase checkpoint is activated by replication fork stalling, and is probably initiated both by the specialized checkpoint sensors (via inactivation of the S phase promoters cyclin E/Cdk2) and by various repair proteins such as the MRN complex (MRE11/RAD50/NBS1) and BRCA1. Activation by the latter group of sensors is thought to also activate a second pathway via phosphorylation of SMC1 (structure and maintenance of chromosomes 1) and SMC3 (structure and maintenance of chromosomes 3), which promotes recombination repair to recover stalled or collapsed replication forks. The G2/M checkpoint, which prevents initiation of mitosis in the presence of DNA damage, operates via the ATM and ATR-mediated to regulate G2/M transition via inactivation of Cdc25C/Cyclin B.Citation14 Maintenance of the G2/M arrest requires transcriptional repression of Cdc2 and Cyclin B expression, mediated via activation of p53 and p21.
Activation of p53 plays an important role in apoptotic signaling. It has been implicated in activation of the intrinsic apoptosis pathway by shifting the balance of the multifunctional B-cell lymphoma-2 (Bcl-2) family away from Bcl-2 survival signaling towards induction of proapoptotic factors such as Bax and PUMA (p53 upregulated modulator of apoptosis), which contribute to caspase cascade activation.Citation76 Furthermore, p53 has been linked to activation of Fas and DR5/KILLER death receptors, which activate the extrinsic apoptosis pathway that also contributes to caspase-mediated cell death.Citation77
Clinical implications of DNA repair in cancer
Alterations in expression of DNA repair may influence cancer biology and influence aggressive phenotypes. Germ-line polymorphism of the POLB gene (rs3136797) encoding a Polβ variant with a low catalytic activity has been recently shown to induce cellular transformation and may be associated with increased cancer susceptibility.Citation78 About 30% of human tumors appear to express Polβ variant proteins (such as K289M or I260M) which can induce cellular transformation in vitro, associated with an aggressive mutator phenotype.Citation79
Overexpression of DNA repair factors may promote cell survival in established tumors. For example, ROS generated during increased metabolic activity in cancer cells generate DNA damaging lesions such as AP sites, oxidative base damage, DNA SSBs, and DNA DSBs. If unrepaired, such DNA lesions could be deleterious to the cancer cell. Moreover, hypoxic and acidic tumor microenvironments can promote further oxidative stress in cancer cells. Although ROS scavenging systems (such as glutathione and thioredoxin, superoxide dismutases, catalases, and peroxidases) do operate in cancer cells, capacity is limited, eventually leading to ROS-induced DNA damage. Therefore cancer cells also utilize the DNA repair machinery to process DNA damaging lesions and maintain cellular survival. Clinical evidence supports the hypothesis that overexpression of DNA repair factors may have prognostic and predictive significance in patients (reviewed in Abbotts and Madhusudan).Citation21
Taken together, this evidence suggests that targeting DNA repair is a valid anticancer strategy. A detailed discussion is beyond the scope of this article, as several recent comprehensive reviews are available.Citation80–Citation83 Here, we focus on the current status of PARP inhibitors in cancer therapy.
The most advanced class of DNA repair inhibitors to date are PARP inhibitors, which disrupt the BER-related SSBR pathway. PARP1 senses and binds to DNA strand breaks, catalyzing (auto-) poly(ADP-ribosyl)ation of target proteins to induce localized chromatin relaxation and assembly of an XRCC1-LIG3-PNKP repair complex. A number of potential PARP inhibitors have been identified, usually with nonspecificity within the PARP family due to high sequence homology at the active site. In vitro and in xenograft models, PARP inhibitors have been demonstrated to potentiate the action of a wide variety of damaging agents, including platinums, the alkylating agents temozolomide and cyclophosphamide, the nucleoside analogue gemcitabine, the topoisomerase inhibitor irinotecan, and ionizing radiation.Citation84,Citation85 Several PARP inhibitors have entered the clinical setting in Phase I–III studies in combination with various chemotherapeutic agents, although results have been mixed (reviewed recently by Davar et al).Citation86 For example, the Pfizer compound rucaparib (AG-014699; Pfizer, Inc., New York, NY, USA) has been evaluated in Phase I and II in combination with temozolomide in malignant melanoma, demonstrating successful PARP inhibition at a tissue level and probable anticancer activity, but significant myelosuppression causing dose-limiting toxicity.Citation87 Similar toxicity has been noted with olaparib (AZD2281; AstraZeneca, London, UK) in combination with paclitaxel, carboplatin, or cisplatin and gemcitabine in Phase III trials in gastric cancer.Citation88 Myelosuppression or other dose-limiting toxicities have not been noted with iniparib (BSI-201; Sanofi, Paris, France), which has been evaluated at Phase II in metastatic triple-negative breast cancer in combination with gemcitabine and carboplatin. A significantly improved median overall survival was demonstrated compared with gemcitabine and carboplatin alone, without increased toxicity. However, a Phase III trial failed to meet co-primary endpoints of overall and progression-free survival,Citation89 and after further disappointing results in a Phase III non-small-cell lung cancer trial, iniparib has been suspended from further development.Citation90 It should be noted that doubts have been raised about iniparib’s ability to inhibit PARP activity. Although initially believed to noncompetitively inhibit PARP1 by association with the DNA binding domain, more recent studies have failed to demonstrate target inhibition.Citation91 A good safety profile was also observed with veliparib (ABT-888; Abbott Laboratories, Abbott Park, IL, USA) in combination with temozolomide. This was associated with positive early results in metastatic colorectal and BRCA-deficient breast cancers, although in advanced melanoma the combination was associated with poor response and no progression-free or overall survival improvement. Many additional Phase I and II trials are currently underway, in combination with a variety of agents, including carboplatin, 5-fluorouracil and oxaliplatin, cisplatin and paclitaxel, topotecan, gemcitabine, and radiotherapy (reviewed in Davar et al).Citation86 Other PARP inhibitors, including orally bioavailable agents, are currently also under Phase I investigation.Citation86,Citation92
The data presented above suggest that the clinical utility of PARP inhibitors in combination with chemotherapy may be limited in tumors in view of narrow therapeutic index. However, evolving preclinical and clinical evidence provides compelling data that DNA repair inhibitor use could be targeted more effectively by utilizing a synthetic lethality strategy.
Synthetic lethality
Synthetic lethality exploits inter-gene relationships where the loss of function of either of two related genes is nonlethal, but loss of both causes cell death. This offers the potential to specifically target cancer cells through inhibition of a gene known to be in a synthetic lethal relationship with a mutated tumor suppressor gene.Citation93
PARP1 inhibition in BRCA deficiency
The best-characterized synthetic lethality relationship is between BRCA mutation and PARP1 inhibition.Citation94–Citation96 PARP1 plays a role in the BER-related pathway of SSBR. Inhibition of SSBR is associated with accumulation of DSBs, which can be exploited in a subset of cancers possessing defects in DSB repair. BRCA1 and −2 have long been identified as tumor suppressors, being mutated in an inherited cancer predisposition that increases susceptibility to breast and ovarian tumors.Citation97 Both BRCA gene products have a role in the HR DNA repair pathway.Citation98 In BRCA-deficient cells, loss of effective HR leads to DSB persistence and cell death (). As heterozygosity at a BRCA allele is associated with effective HR, DSB accumulation induced by PARP inhibition specifically occurs only in tumor cells with acquired BRCA−/− homozygosity.Citation7,Citation8 Furthermore, loss of BRCA2 function has been linked to hyperactivation of PARP1 (an observation replicated in loss of other HR factors), enhancing the cytotoxic effect.Citation99
Figure 2 Synthetic lethality in BRCA−/− cells upon PARP inhibition. PARP inhibition leads to SSBs. During replication SSBs get converted to DSBs. BRCA−/− cells are deficient in HR and hence unable to repair DSBs. DSB accumulation leads to cell death. In cells where PARP is proficient, SSBs are repaired by BER irrespective of BRCA status. There is no DSB generation, and cells continue to survive.
Abbreviations: BER, base excision repair; BRCA, breast cancer susceptibility protein; DSB, double-strand break; HR, homologous recombination; PARP, poly[ADP-ribose] polymerase 1; SSB, single-strand break.
![Figure 2 Synthetic lethality in BRCA−/− cells upon PARP inhibition. PARP inhibition leads to SSBs. During replication SSBs get converted to DSBs. BRCA−/− cells are deficient in HR and hence unable to repair DSBs. DSB accumulation leads to cell death. In cells where PARP is proficient, SSBs are repaired by BER irrespective of BRCA status. There is no DSB generation, and cells continue to survive.Abbreviations: BER, base excision repair; BRCA, breast cancer susceptibility protein; DSB, double-strand break; HR, homologous recombination; PARP, poly[ADP-ribose] polymerase 1; SSB, single-strand break.](/cms/asset/864256f5-d4ee-42ca-9418-1bc65ae3b84e/dcmr_a_50497_f0002_c.jpg)
PARP inhibition has been hypothesized to cause persistence of endogenously generated SSBs, inducing collapse of replication forks and formation of lethal DSBs.Citation100 However, the exact mechanism of PARP inhibition has not been fully elucidated. Because of its role in the related SSBR pathway, PARP1 has occasionally and erroneously been described as essential for BER, although knockdown models have demonstrated this to be inaccurate. Unlike many BER factors, PARP1 is not essential for viability, nor is it required for repair of BER substrates such as alkylation damage – in actuality, PARP1 has been demonstrated to reduce BER kinetics.Citation101 Interestingly, the mode of PARP1 inactivation may impact the biological consequences. For example, small interfering RNA (siRNA)-mediated PARP1 knockdown does not induce significant cytotoxicity in BRCA-deficient cells,Citation7 while small molecule inhibition induces SSB accumulation after alkylating agent treatment and is well documented to be synthetically lethal in BRCA-deficient cells.Citation101 This is probably because the BER intermediate single-strand nick is a substrate for transient PARP1 binding, hence accounting for slowed BER kinetics in the presence of PARP1. In this model, PARP inhibitor binding may trap PARP1 onto an SSB, whether formed spontaneously, exogenously, or by BER.Citation102 As a result, downstream repair (whether by SSBR or BER) is prevented, leading to toxic DSBs during replication.Citation103 Phase I and II trials of PARP inhibitors have demonstrated favorable efficacy and limited toxicity in BRCA-related breast and ovarian cancers.Citation104 An initial Phase I study of olaparib in a cohort enriched for BRCA1/2 mutation carriers demonstrated evidence of in vivo anti-PARP activity (using PARP activity measurement in peripheral blood mononuclear cells and the surrogate marker of γH2AX induction, which accumulates at DSBs) and evidence of response in 40% BRCA carriers.Citation96 This led to Phase II trials in breast or ovarian cancer associated with BRCA1/2 mutation, demonstrating further favorable data suggesting antitumor efficacy in this cohort.Citation105,Citation106 Phase III trials in BRCA-mutated ovarian cancer are currently planned.Citation107,Citation108 Likewise, Phase II investigation of rucaparib in BRCA1/2-mutated breast or ovarian cancer demonstrates PARP activity inhibition and evidence of tumor response.Citation109 The oral PARP1/2 inhibitor niraparib (MK4827; Merck & Co, Inc., Whitehouse Station, NJ, USA) has also been evaluated at Phase I to possess an acceptable safety profile and probable antitumor activity.Citation110 “BRCAness” refers to a subset of breast cancers, including “triple negative” (estrogen-, progesterone-, and HER2 [human epidermal growth factor receptor 2]-negative) and “basal phenotype” cancers, that possess molecular and histopathological similarity to BRCA-deficient tumors, which have been successfully targeted in vitro by PARP inhibition.Citation111,Citation112 Similarly, high-grade serous/undifferentiated ovarian cancers (HGSOC) are commonly associated with somatic or epigenetic loss of BRCA1/2.Citation113 Phase II investigations of olaparib in these cohorts initially suggested that there may be a role for PARP inhibition in HGSOC,Citation114 although Phase III development was terminated after it was deemed unlikely that reported progression-free survival would translate to an overall benefit.Citation115 It should be noted that BRCA deficiency does not translate to PARP inhibitor response in all patients, and resistance may be a significant problem in future development. Two groups independently described the deletion of a previously identified BRCA2 mutation in PARP inhibitor-resistant cancer cells that led to restoration of the open reading frame, and hence HR proficiency.Citation116,Citation117 Furthermore, in BRCA1-deleted cells, loss of expression of the HR protein 53BP1 appears to partially restore HR competency and abrogate the ATM-dependent checkpoint response, limiting the resultant cell cycle arrest triggered by DSB accumulation after DNA damaging agent exposure.Citation118
Alternative synthetic lethality partners for BER
The discovery of the synthetic lethality relationship between PARP1 and BRCA suggests that other tumor-specific defects in DSB repair factors may be therapeutically targeted by PARP inhibition. Germline mutations in the HR protein RAD51D have been identified as conferring susceptibility to ovarian cancer and may offer a target for PARP inhibitors in a small subset of women.Citation119 Recent evidence suggests single agent cytotoxicity of PARP inhibitors in cells with reduced expression of ATM, the checkpoint activator that is activated by DSBs.Citation120,Citation121 Similar results have been observed in cells deficient in expression of the HR protein MRE11,Citation122 and following in vitro downregulation of Artemis or LIG4, both of which function within the NHEJ pathway.Citation123,Citation124 Other potential synthetic lethality partners in PARP inhibition identified on a high-throughput siRNA screen include the DSB-induced checkpoint activator ATR, and a variety of factors that have been associated with bypass of stalled replication or transcription forks, including PCNA, DDB1, and XAB2 (XPA-binding protein 2).Citation125 Conversely, SSBR factors other than PARP1 are potential synthetic lethality partners in DSB repair loss, as observed by the cytotoxicity induced by inhibitors of ATM or DNA-PKcs following knockdown of the BER protein XRCC1.Citation126 Given its critical role in BER, targeting APE1 also presents a promising alternative. Sultana et alCitation121 recently demonstrated that novel small molecule APE1 inhibitors are able to induce AP site accumulation, DSBs, cell cycle arrest, and cytotoxicity in BRCA2- or ATM-deficient cells. The synthetic lethality relationship between HR and APE1 was confirmed by cytotoxicity observed following ATM inhibitor exposure in APE1−/− cells.Citation121
In addition to targeting tumors harboring germline defects in HR, Seo et al highlighted a possible treatment strategy in sporadic tumors by demonstrating that inhibition of APE1 DNA repair function induces targeted cytotoxicity in cell lines cultured in acidic environments.Citation127 Tumor microenvironments are commonly acidic and have been associated with upregulation of BER proteins, including APE1. Conversely, other DNA repair mechanisms, including HR, are often downregulated under such conditions.Citation128,Citation129 Identification of tumors with BER upregulation and HR depletion may therefore offer an opportunity to exploit synthetic lethality through APE1 inhibition.
Recent evidence suggests that relationships between BER and non-HR DNA repair pathways may hold potential for synthetic lethality. For example, 8-oxoguanine base lesions, which are induced by metabolic ROS and can cause mutagenic GC→TA transversions if unrepaired, may be processed by both BER and MMR Mutations in the MMR genes MLH1 or MSH2 are implicated in HNPCC and some sporadic colorectal cancers. SiRNA inhibition of the BER constituent DNA polymerases β/γ have been demonstrated to be selectively lethal in MLH1/MSH2 mutant cell lines, suggesting a synthetic lethality relationship.Citation130 It remains to be established whether additional factors such as APE1 may have a role in this capacity.
Phosphatase and tensin homolog mutation as a synthetic lethality target
Phosphatase and tensin homolog (PTEN) is a negative regulator of the anti-apoptotic PI3K/Akt pathway. In addition to its inositol phosphatase function, PTEN has recently been implicated in the maintenance of genomic integrity.Citation131–Citation136 On the basis of evidence supporting an HR defect associated with PTEN mutation, Mendes-Pereira et alCitation38 tested for synthetic lethality in HCT116 colorectal tumor cells transfected with a PTEN-mutant cDNA clone. Homozygosity for PTEN mutation was associated with a 20-fold increase in sensitivity to PARP inhibitors, which was replicated in a panel of commonly cultured tumor cell lines and in mouse xenografts. Ectopic expression of wild type PTEN into a PTEN-deficient prostate cancer cell line abrogated this effect, as did induced expression of a PTEN phosphatase domain mutant, suggesting that PTEN’s influence over HR lies out with its phosphatase function. Ectopic expression of RAD51 in a PTEN-deficient cell line was also able to overcome PARP inhibitor sensitivity, supporting the proposed link between PTEN mutation and reduced RAD51 expression. Similar results were demonstrated in endometrioid endometrial carcinoma, in which PTEN is mutated in up to 80% of patients.Citation137 In primary PTEN−/− mouse astrocytes, reduced transcription of the RAD51 paralogs was associated with sensitivity to PARP inhibition,Citation138 while PTEN disruption in colorectal cancer cells resulted in reduced MRE11 accumulation at DSBs that is also associated with PARP inhibitor sensitivity.Citation139 In lung cancer cells, PTEN deficiency potentiated the synergistic effect of olaparib and cisplatin combination treatment,Citation140 while rucaparib sensitized PTEN-deficient prostate cancer cells to ionizing radiation,Citation141 with both reports highlighting delayed DSB repair kinetics as a likely mechanism.
In the clinical setting, there is anecdotal evidence of successful targeting of PTEN deficiency with PARP inhibitor. Forster et alCitation142 have presented a case study of usage of the PARP inhibitor olaparib in a patient with platinum- responsive metastatic endometrioid endometrial adenocarcinoma. Treatment was initiated following development of brain metastases, on the basis of previous sensitivity with platinum agents, which are highly effective in HR deficiency. A partial response on magnetic resonance imaging was noted at 10 weeks, followed by a progression-free survival of 8 months. Tumor biopsy demonstrated PTEN mutation with wild type BRCA1 and −2 status.Citation142
However, there is not a consensus regarding the synthetic lethality relationship between PTEN deficiency and PARP inhibition. In studies on PTEN-null prostateCitation139 and lungCitation143 cancer cells, no enhanced DNA damaging agent or PARP inhibitor sensitivity was observed. Furthermore, in a Phase I trial in BRCA mutation carriers and sporadic cancer, PTEN status did not correlate with antitumor activity of niraparib.Citation144
Although PARP inhibitor-induced synthetic lethality in PTEN loss has been most widely studied, a recent report from Mereniuk et alCitation145 provides evidence that other proteins within the SSBR pathways may also be valid targets. A forward transfection screen of nearly 7,000 siRNAs was performed using A549 lung cancer cells stably depleted of the BER end-processing enzyme PNKP. This screen identified PTEN as a potential synthetic lethal partner, a result then validated by: 1) repeat siRNA downregulation of PTEN in PNKP-null MCF7 (Michigan Cancer Foundation-7) breast cancer cells; and 2) PNKP inhibitor exposure in PTEN-null HCT116 (colorectal) and PC3 (prostate) cancer cells. Furthermore, PNKP inhibitor treatment in PTEN-deficient cells is associated with accumulation of DSBs, increased apoptosis, and reduced clonogenic survival in a manner analogous to published reports of PARP inhibition in BRCA mutation. The authors hypothesize that PTEN loss is associated with strand-break accumulation that may require PNKP-mediated end processing for repair. Although a mechanism for strand breakage is not presented, sensitivity to PNKP inhibitor could not be abrogated by ectopic RAD51 expression in PTEN-null PC3 cells, in keeping with previous reports that synthetic lethality in this cell line is not mediated via RAD51 loss.
Developing biomarkers for synthetic lethality response
BRCA mutation, although an excellent marker of HR deficiency, comprises a small subset of breast and ovarian patients. Identification of other synthetic lethality relationships is ongoing, as described above. A number of individual DNA repair proteins may therefore be informative regarding HR deficiency and synthetic lethality response (reviewed in Martin et al).Citation130 An alternative approach to identify patients who may benefit from PARP (or BER) inhibition is to use gene expression profiles that predict responsiveness. While data regarding PARP inhibitor response is currently limited, there is a large body of evidence related to anthracycline or platinum sensitivity. As both classes are associated with DSB induction (via intercalation/topoisomerase inhibition and crosslink formation, respectively), it may be predicted that response to such agents would also equate to PARP inhibitor response. By correlating DNA repair gene expression microarray data with anthracycline sensitivity in triple-negative breast cancerCitation146 and platinum sensitivity in epithelial ovarian cancer,Citation147 two groups have been able to develop “BRCAness gene signatures” which reproducibly predict treatment response. In the ovarian cancer study, this was further analyzed in BRCA2-mutated pancreatic cancer cell clones to predict for RAD51 foci formation as a marker of HR, and for sensitivity to PARP inhibition. Independently, RAD51 foci formation in tumor samples has been developed as a functional assay for HR status.Citation148 Primary cultures of epithelial ovarian cancer cells from ascitic fluid can be assessed for γH2AX and RAD51 foci, which correlate with in vitro response to the PARP inhibitor.
Assays have also been developed for monitoring effective PARP inhibition on treatment. In multiple clinical trials, PARP activity in peripheral mononuclear blood cells (measured as cellular levels of poly[ADP]-ribose polymers detected by immunofluorescence or enzyme-linked immunosorbent assay) has been used as a marker of effective inhibition.Citation149,Citation150 Surrogate markers, such as comet assay assessment of DNA damage level, or DSB estimation by RAD51 or γH2AX foci after treatment, have also been used.Citation151 However, it is important to note that Phase II studies of olaparib have indicated that the maximum tolerated dose, determined by conventional dose escalation, may induce a better clinical response than the lowest effective PARP inhibitory dose.Citation105,Citation106
Conclusion
DNA repair mechanisms play an essential role in promoting genomic stability. Defective DNA repair may predispose to cancer. On the other hand, impaired DNA repair capacity in cancer cells may influence a favorable response to chemotherapy and radiotherapy. Recent evidence demonstrates that overexpression of DNA repair factors has prognostic and predictive significance in cancer patients. More recently, DNA repair has emerged as a new area for anticancer drug discovery. Use of DNA repair inhibitors in combination with chemotherapy or radiotherapy can increase cancer cell killing, although combination strategies can lead to profound normal tissue toxicity. The strategy of synthetic lethality to exploit interrelationships between DNA repair pathways appears to bypass many problems associated with combination strategies. The recent success of PARP inhibitors in BRCA-deficient breast and ovarian cancer clearly suggests that additional factors within DNA repair are likely to be promising synthetic lethality targets in the future and have the potential to transform the therapeutic landscape in cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
- HoeijmakersJHGenome maintenance mechanisms for preventing cancerNature2001411683536637411357144
- SweasyJBLangTDiMaioDIs base excision repair a tumor suppressor mechanism?Cell Cycle20065325025916418580
- GoodeELUlrichCMPotterJDPolymorphisms in DNA repair genes and associations with cancer riskCancer Epidemiol Biomarkers Prev200211121513153012496039
- FrosinaGCommentary: DNA base excision repair defects in human pathologiesFree Radic Res200438101037105415512792
- LongleyDBJohnstonPGMolecular mechanisms of drug resistanceJ Pathol2005205227529215641020
- MadhusudanSHicksonIDDNA repair inhibition: a selective tumour targeting strategyTrends Mol Med2005111150351116214418
- FarmerHMcCabeNLordCJTargeting the DNA repair defect in BRCA mutant cells as a therapeutic strategyNature2005434703591792115829967
- BryantHESchultzNThomasHDSpecific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymeraseNature2005434703591391715829966
- LoebLASpringgateCFBattulaNErrors in DNA replication as a basis of malignant changesCancer Res1974349231123214136142
- LambrosMBNatrajanRGeyerFCPPM1D gene amplification and overexpression in breast cancer: a qRT-PCR and chromogenic in situ hybridization studyMod Pathol201023101334134520543821
- EkerAPQuayleCChavesIvan der HorstGTDNA repair in mammalian cells: direct DNA damage reversal: elegant solutions for nasty problemsCell Mol Life Sci200966696898019153659
- LordCJAshworthABiology-driven cancer drug development: back to the futureBMC Biol201083820385032
- NatrajanRWeigeltBMackayAAn integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal-like, HER2 and luminal cancersBreast Cancer Res Treat2010121357558919688261
- SancarALindsey-BoltzLAUnsal-KacmazKLinnSMolecular mechanisms of mammalian DNA repair and the DNA damage checkpointsAnnu Rev Biochem200473398515189136
- FortiniPPascucciBParlantiED’ErricoMSimonelliVDogliottiEThe base excision repair: mechanisms and its relevance for cancer susceptibilityBiochimie200385111053107114726013
- NilsenHKrokanHEBase excision repair in a network of defence and toleranceCarcinogenesis200122798799811408341
- IzumiTWiederholdLRRoyGMammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damageToxicology20031931–2436514599767
- DianovGAllinsonSLBudworthHSleethKMMammalian base excision repairCaldecottKWEukaryotic DNA Damage Surveillance and RepairKluwer Academic/Plenium Publishers2003122
- BarnesDELindahlTRepair and genetic consequences of endogenous DNA base damage in mammalian cellsAnnu Rev Genet20043844547615568983
- RobertsonABKlunglandARognesTLeirosIDNA repair in mammalian cells: base excision repair: the long and short of itCell Mol Life Sci200966698199319153658
- AbbottsRMadhusudanSHuman AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancerCancer Treat Rev201036542543520056333
- WilsonDM3rdKimDBerquistBRSigurdsonAJVariation in base excision repair capacityMutat Res20117111–210011221167187
- WangJCCellular roles of DNA topoisomerases: a molecular perspectiveNat Rev Mol Cell Biol20023643044012042765
- PommierYRedonCRaoVARepair of and checkpoint response to topoisomerase I-mediated DNA damageMutat Res20035321–217320314643436
- CaldecottKWSingle-strand break repair and genetic diseaseNat Rev Genet20089861963118626472
- ParlantiELocatelliGMagaGDogliottiEHuman base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteinsNucleic Acids Res20073551569157717289756
- TanYRaychaudhuriPCostaRHChk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genesMol Cell Biol20072731007101617101782
- ChenDYuZZhuZLopezCDE2F1 regulates the base excision repair gene XRCC1 and promotes DNA repairJ Biol Chem200828322153811538918348985
- SugasawaKRegulation of damage recognition in mammalian global genomic nucleotide excision repairMutat Res20106851–2293719682467
- ShuckSCShortEATurchiJJEukaryotic nucleotide excision repair: from understanding mechanisms to influencing biologyCell Res2008181647218166981
- de BoerJHoeijmakersJHNucleotide excision repair and human syndromesCarcinogenesis200021345346010688865
- IornsELordCJGrigoriadisAIntegrated functional, gene expression and genomic analysis for the identification of cancer targetsPLoS One200944e512019357772
- SourisseauTManiotisDMcCarthyAAurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibitionEmbo Mol Med20102413014220373286
- LiGMMechanisms and functions of DNA mismatch repairCell Res2008181859818157157
- HewishMLordCJMartinSACunninghamDAshworthAMismatch repair deficient colorectal cancer in the era of personalized treatmentNat Rev Clin Oncol20107419720820177404
- MartinSALordCJAshworthATherapeutic targeting of the DNA mismatch repair pathwayClin Cancer Res201016215107511320823149
- MartinSAHewishMLordCJAshworthAGenomic instability and the selection of treatments for cancerJ Pathol2010220228128919890832
- Mendes-PereiraAMMartinSABroughRSynthetic lethal targeting of PTEN mutant cells with PARP inhibitorsEmbo Mol Med200916–731532220049735
- MahaneyBLMeekKLees-MillerSPRepair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joiningBiochem J2009417363965019133841
- KassEMJasinMCollaboration and competition between DNA doublestrand break repair pathwaysFEBS Lett2010584173703370820691183
- MladenovEIliakisGInduction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathwaysMutat Res20117111–2617221329706
- McVeyMLeeSEMMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endingsTrends Genet2008241152953818809224
- de VillartayJPV(D)J recombination deficienciesAdv Exp Med Biol2009650465819731800
- NussenzweigANussenzweigMCA backup DNA repair pathway moves to the forefrontCell2007131222322517956720
- KatsuraYSasakiSSatoMInvolvement of Ku80 in microhomology-mediated end joining for DNA double-strand breaks in vivoDNA Repair (Amst)20076563964817236818
- SchiplerAIliakisGDNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choiceNucleic Acids Res201341167589760523804754
- KavanaghJNRedmondKMSchettinoGPriseKMDNA double strand break repair: a radiation perspectiveAntioxid Redox Signal201318182458247223311752
- ShiLOberdoerfferPChromatin dynamics in DNA double-strand break repairBiochim Biophys Acta20121819781181922285574
- MurrayJMStiffTJeggoPADNA double-strand break repair within heterochromatic regionsBiochem Soc Trans201240117317822260685
- LieberMRThe mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathwayAnnu Rev Biochem20107918121120192759
- KobayashiJIwabuchiKMiyagawaKCurrent topics in DNA double-strand break repairJ Radiat Res20084929310318285658
- HelledayTLoJvan GentDCEngelwardBPDNA double-strand break repair: from mechanistic understanding to cancer treatmentDNA Repair (Amst)20076792393517363343
- WymanCKanaarRDNA double-strand break repair: all’s well that ends wellAnnu Rev Genet20064036338316895466
- CahillDConnorBCarneyJPMechanisms of eukaryotic DNA double strand break repairFront Biosci2006111958197616368571
- van den BoschMLohmanPHPastinkADNA double-strand break repair by homologous recombinationBiol Chem2002383687389212222678
- KarranPDNA double strand break repair in mammalian cellsCurr Opin Genet Dev200010214415010753787
- FeatherstoneCJacksonSPDNA double-strand break repairCurr Biol1999920R759R76110531043
- HanadaKIkedaH[Double strand break repair via DNA end-joining]Tanpakushitsu Kakusan Koso199944Suppl 1218381844 Japanese10503021
- KanaarRHoeijmakersJHvan GentDCMolecular mechanisms of DNA double strand break repairTrends Cell Biol19988124834899861670
- TsukamotoYIkedaHDouble-strand break repair mediated by DNA end-joiningGenes Cells1998331351449619626
- SargentRGBrennemanMAWilsonJHRepair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombinationMol Cell Biol19971712672778972207
- Saleh-GohariNBryantHESchultzNParkerKMCasselTNHelledayTSpontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaksMol Cell Biol200525167158716916055725
- LawleyPDPhillipsDHDNA adducts from chemotherapeutic agentsMutat Res19963551–213408781575
- BoglioloMSchusterBStoepkerCMutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemiaAm J Hum Genet201392580080623623386
- KumaresanKRSridharanDMMcMahonLWLambertMWDeficiency in incisions produced by XPF at the site of a DNA interstrand cross-link in Fanconi anemia cellsBiochemistry20074650143591436818020456
- HoTVScharerODTranslesion DNA synthesis polymerases in DNA interstrand crosslink repairEnviron Mol Mutagen201051655256620658647
- McHughPJSpanswickVJHartleyJARepair of DNA interstrand crosslinks: molecular mechanisms and clinical relevanceLancet Oncol20012848349011905724
- HinzJMRole of homologous recombination in DNA interstrand crosslink repairEnviron Mol Mutagen201051658260320658649
- DeansAJWestSCDNA interstrand crosslink repair and cancerNat Rev Cancer201111746748021701511
- LongDTRaschleMJoukovVWalterJCMechanism of RAD51-dependent DNA interstrand cross-link repairScience20113336038848721719678
- NicolayNHHelledayTSharmaRABiological relevance of DNA polymerase beta and translesion synthesis polymerases to cancer and its treatmentCurr Mol Pharmacol201251546722122464
- YamanakaKLloydRSFunctions of translesion DNA polymerases: implications for cancer risk and opportunities as therapeutic targetsMadhusudanSWilsonDM3rdDNA Repair and Cancer: from Bench to ClinicBoca Raton, USACRC Press2013
- McCullochSDKunkelTAThe fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerasesCell Res200818114816118166979
- HigginsNPKatoKStraussBA model for replication repair in mammalian cellsJ Mol Biol197610134174251255724
- LongDTKreuzerKNFork regression is an active helicase-driven pathway in bacteriophage T4Embo Rep200910439439919270717
- HauptSBergerMGoldbergZHauptYApoptosis – the p53 networkJ Cell Sci2003116Pt 204077408512972501
- PietenpolJAStewartZACell cycle checkpoint signaling: cell cycle arrest versus apoptosisToxicol2002181182475481
- YamtichJNemecAAKehASweasyJBA germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformationPLoS Genet2012811e100305223144635
- StarcevicDDalalSSweasyJBIs there a link between DNA polymerase beta and cancer?Cell Cycle200438998100115280658
- BapatAFishelMKelleyMRGoing ape as an approach to cancer therapeuticsAntioxid Redox Signal200911365166818715143
- BasuBYapTAMolifeLRde BonoJSTargeting the DNA damage response in oncology: past, present and future perspectivesCurr Opin Oncol201224331632422476188
- Megnin-ChanetFBolletMAHallJTargeting poly(ADP-ribose) polymerase activity for cancer therapyCell Mol Life Sci201067213649366220725763
- MizuaraiSKotaniHSynthetic lethal interactions for the development of cancer therapeutics: biological and methodological advancementsHum Genet2010128656757520976469
- DonawhoCKLuoYLuoYABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA- damaging agents in preclinical tumor modelsClin Cancer Res20071392728273717473206
- OssovskayaVLiLBradleyCShermanBAbstract 2311: activity of BSI-201, a potent poly(ADP-ribose) polymerase (PARP1) inhibitor, alone and in combination with topotecan in human ovarian xenografts99th AACR Annual Meeting2008San Diego, CA
- DavarDBeumerJHHamiehLTawbiHRole of PARP inhibitors in cancer biology and therapyCurr Med Chem201219233907392122788767
- PlummerRLoriganPEvansJFirst and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM). In: ASCO Annual Meeting. Atlanta, GAJ Clin Oncol200624 Abstract 8013
- AstraZenecaEfficacy and safety study of olaparib in combination with paclitaxel to treat advanced gastric cancer Available from: http://www.clinicaltrials.gov/ct2/show/NCT01924533. NLM identifier: NCT01924533Accessed August 26, 2013
- Sanofi-aventisSanofi-aventis reports top-line results from phase III study with BSI-201 in metastatic triple-negative breast cancer [press release]Paris, FranceSanofi-aventis1272011 Available from: http://en.sanofi.com/Images/13666_20110127_BSI_en.pdfAccessed August 26, 2013
- Sanofi-aventisSanofi provides update on phase 3 studies of two investigational compounds [press release]Paris, FranceSanofi-aventis632013 Available from: http://en.sanofi.com/Images/33127_20130603_rdupdate_en.pdfAccessed August 26, 2013
- LiuXShiYMaagDXIniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitorClin Cancer Res201218251052322128301
- GlendenningJTuttAPARP inhibitors – current status and the walk towards early breast cancerBreast201120Suppl 3S12S1922015278
- RehmanFLLordCJAshworthASynthetic lethal approaches to breast cancer therapyNat Rev Clin Oncol201071271872420956981
- TumaRSCombining carefully selected drug, patient genetics may lead to total tumor deathJ Natl Cancer Inst2007992015051506150917925530
- LordCJAshworthATargeted therapy for cancer using PARP inhibitorsCurr Opin Phamacol200884363369
- FongPCBossDSYapTAInhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriersN Engl J Med2009361212313419553641
- MikiYSwensenJShattuckeidensDA strong candidate for the breast and ovarian-cancer susceptibility gene BRCA1Science1994266518266717545954
- VenkitaramanARCancer susceptibility and the functions of BRCA1 and BRCA2Cell2002108217118211832208
- GottipatiPVischioniBSchultzNPoly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cellsCancer Res201070135389539820551068
- DurkaczBWOmidijiOGrayDAShallS(ADP-ribose)n participates in DNA excision repairNature198028357475935966243744
- StromCEJohanssonFUhlenMSzigyartoCAErixonKHelledayTPoly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediateNucleic Acids Res20113983166317521183466
- MuraiJHuangSYDasBBTrapping of PARP1 and PARP2 by clinical PARP inhibitorsCancer Res201272215588559923118055
- HelledayTThe underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandingsMol Oncol20115438739321821475
- BalmanaJDomchekSMTuttAGarberJEStumbling blocks on the path to personalized medicine in breast cancer: the case of PARP inhibitors for BRCA1/2-associated cancersCancer Discov201111293422586318
- TuttARobsonMGarberJEOral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trialLancet2010376973723524420609467
- AudehMWCarmichaelJPensonRTOral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trialLancet2010376973724525120609468
- AstraZenecaOlaparib monotherapy in patients with BRCA mutated ovarian cancer following first line platinum based chemotherapy Available from: http://www.clinicaltrials.gov/ct2/show/NCT01844986. NLM identifier: NCT01844986Accessed June 14, 2013
- AstraZenecaOlaparib treatment in BRCA mutated ovarian cancer patients after complete or partial response to platinum chemotherapy Available from: http://www.clinicaltrials.gov/ct2/show/NCT01874353. NLM identifier: NCT01874353Accessed August 26, 2013
- DrewYLedermannJAJonesAAbstract 3104: Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA 1 and 2–mutated, advanced ovarian and/or locally advanced or metastatic breast cancer. ASCO Annual Meeting. Chicago, ILJ Clin Oncol2011293104
- SchelmanWRSandhuSKMorenoGarcia VFirst-in-human trial of a poly(ADP)-ribose polymerase (PARP) inhibitor MK-4827 in advanced cancer patients with antitumor activity in BRCA-deficient tumors and sporadic ovarian cancers (SOC). ASCO Annual Meeting. Chicago, ILJ Clin Oncol201129 Abstract 3102
- TurnerNTuttAAshworthAHallmarks of ‘BRCAness’ in sporadic cancersNat Rev Cancer200441081481915510162
- GiorgettiGGaliziaEBianchiFBrcaness phenotype and methylation of BRCA1 promoter in sporadic breast cancersAnn Oncol200718525217047001
- HennessyBTTimmsKMCareyMSSomatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancerJ Clin Oncol201028223570357620606085
- GelmonKATischkowitzMMackayHOlaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised studyLancet Oncol201112985286121862407
- AstraZenecaAstraZeneca updates on olaparib and TC-5214 development programmes [press release]London, UKAstraZeneca12202011 Available from: http://www.astrazeneca.com/Media/Press-releases/Article/20111220-az-updates-olaparib-TC5214-developmentAccessed August 29, 2012
- EdwardsSLBroughRLordCJResistance to therapy caused by intragenic deletion in BRCA2Nature200845171821111111518264088
- SakaiWSwisherEMKarlanBYSecondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancersNature200845171821116112018264087
- BouwmanPAlyAEscandellJM53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancersNat Struct Mol Biol201017668869520453858
- LovedayCTurnbullCRamsayEGermline mutations in RAD51D confer susceptibility to ovarian cancerNat Genet201143987988221822267
- WilliamsonCTKubotaEHamillJDEnhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53Embo Mol Med20124651552722416035
- SultanaRMcNeillDRAbbottsRSynthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitorsInt J Cancer2012131102433244422377908
- VilarEBartnikCMStenzelSLMRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancersCancer Res20117172632264221300766
- KurosawaASaitoSSoSDNA ligase IV and artemis act cooperatively to suppress homologous recombination in human cells: implications for DNA double-strand break repairPLoS One201388e7225323967291
- LoserDAShibataAShibataAKWoodbineLJJeggoPAChalmersAJSensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repairMol Cancer Ther2010961775178720530711
- LordCJMcDonaldSSwiftSTurnerNCAshworthAA high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivityDNA Repair (Amst)20087122010201918832051
- SultanaRAbdel-FatahTAbbottsRTargeting XRCC1 deficiency in breast cancer for personalized therapyCancer Res20137351621163423253910
- SeoYKinsellaTJEssential role of DNA base excision repair on survival in an acidic tumor microenvironmentCancer Res200969187285729319723658
- BindraRSGibsonSLMengAHypoxia-induced down-regulation of BRCA1 expression by E2FsCancer Res20056524115971160416357170
- BindraRSSchafferPJMengAAlterations in DNA repair gene expression under hypoxia: elucidating the mechanisms of hypoxia-induced genetic instabilityAnn N Y Acad Sci2005105918419516382054
- MartinSAMcCabeNMullarkeyMDNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1Cancer Cell201017323524820227038
- GuptaAYangQPanditaRKCell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repairCell Cycle20098142198221019502790
- PucJKeniryMLiHSLack of PTEN sequesters CHK1 and initiates genetic instabilityCancer Cell20057219320415710331
- ShenWHBalajeeASWangJLEssential role for nuclear PTEN in maintaining chromosomal integrityCell2007128115717017218262
- MingMHeYYPTEN in DNA damage repairCancer Lett2013319212512922266095
- YinYShenWHPTEN: a new guardian of the genomeOncogene200827415443545318794879
- PlanchonSMWaiteKAEngCThe nuclear affairs of PTENJ Cell Sci2008121Pt 324925318216329
- DedesKJWetterskogDMendes-PereiraAMPTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitorsSci Transl Med201025353ra75
- McEllinBCamachoCVMukherjeeBPTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitorsCancer Res201070135457546420530668
- FraserMZhaoHLuotoKRPTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapyClin Cancer Res20121841015102722114138
- MinamiDTakigawaNTakedaHSynergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cellsMol Cancer Res201311214014823239809
- ChatterjeePChoudharyGSSharmaAPARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cellsPLoS One201384e6040823565244
- ForsterMDDedesKJSandhuSTreatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancerNat Rev Clin Oncol20118530230621468130
- PappasGZumsteinLAMunshiAHobbsMMeynREAdenoviral-mediated PTEN expression radiosensitizes non-small cell lung cancer cells by suppressing DNA repair capacityCancer Gene Ther200714654354917431403
- SandhuSKSchelmanWRWildingGThe poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trialLancet Oncol201314988289223810788
- MereniukTREl GendyMAMendes-PereiraAMSynthetic lethal targeting of PTEN-deficient cancer cells using selective disruption of polynucleotide kinase/phosphataseMol Cancer Ther201312102135214423883586
- RodriguezAAMakrisAWuMFDNA repair signature is associated with anthracycline response in triple negative breast cancer patientsBreast Cancer Res Treat2010123118919620582464
- KonstantinopoulosPASpentzosDKarlanBYGene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancerJ Clin Oncol201028223555356120547991
- MukhopadhyayAElattarACerbinskaiteADevelopment of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitorsClin Cancer Res20101682344235120371688
- MenearKAAdcockCBoulterR4-[3-(4-cyclopropanecar-bonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1J Med Chem200851206581659118800822
- LubbersLSRoweBAHodgeLMPISA, a novel pharmacodynamic assay for assessing poly(ADP-ribose) polymerase (PARP) activity in situJ Pharmacol Toxicol Methods201061331932820132901
- PlummerRJonesCMiddletonMPhase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumorsClin Cancer Res200814237917792319047122