
Abstract
Background
The debate surrounding the acceptance of progression-free survival (PFS) as an intermediate endpoint to overall survival (OS) has grown in recent years, due to the challenges in demonstrating an OS benefit within clinical trials today. PFS is generally a good predictor of OS for cases where survival post-progression (SPP) is short, and less so when SPP is long. SPP depends on multiple factors, including residual effect from experimental treatment and effect from crossover or other subsequent therapies, posing unique challenges into the translation of PFS benefit into OS.
Methods
The objective of this analysis was to conduct simulations investigating how increasing SPP impacts PFS translation to OS, utilizing data from the AXIS (axitinib versus sorafenib in advanced metastatic renal cell carcinoma) trial. The underlying assumption was a treatment benefit in PFS (the PFS distribution parameters were chosen to be equal to median PFS in the AXIS trial) but no treatment effect on SPP, implying that PFS improvement is directly reflected in OS improvement.
Results
The probability of a statistically significant difference between arms for OS decreased from 54.7% to 6.1% when median SPP was increased from one to 20 months. The probability of the hazard ratio of OS being ≥0.9 was similarly increased from 24.3% to 72.6%, even though the hazard ratio for PFS was 0.69.
Conclusion
The present study shows that when simulated SPP is added to trial PFS data, the existing PFS benefit is diluted. Knowing that the AXIS treatment arms are well balanced with respect to post-trial treatments, we conclude that the PFS to OS benefit translation is primarily obscured by random variability largely unrelated to the true outcomes. The implications for drug development are not insignificant, as there would be a need to include more patients in studies or utilize a longer follow-up time to overcome the SPP variability issue.
Introduction
Overall survival (OS), ie, time elapsed between randomization and death from any cause, has historically been the gold standard for demonstrating clinical benefit for oncology treatment.Citation1,Citation2 OS is an attractive endpoint due to its obvious clinical relevance, unambiguous definition, and ease of measurement that is not subject to assessment bias. However, when assessing an OS difference among treatment alternatives, the results can be impacted by nonsystematic use of subsequent therapy that patients may go on to receive.Citation1,Citation3–Citation5
Assessing OS, especially in cancers where multiple therapies are available, may require longer timelines and greater cost if more patients need to be enrolled to achieve the required number of events sooner, and a much lengthier follow-up period to document the events. In particular, in a head-to-head clinical trial where both the new regimen and current standard of care include treatments that provide long survival, insistence on proven OS benefits for new drugs may delay availability and timely development of new efficacious treatments for patients.Citation6
Progression-free survival (PFS) is defined as the time elapsed between randomization and tumor progression or death from any cause, with censoring of patients without an event at their last set of lesion measurements verifying lack of progression. PFS is arguably the preferred choice among alternatives to OS, in that it is accessible early, and requires smaller patient samples and shorter follow-up for a desired power;Citation2,Citation3 however, it also has some important limitations. Unlike OS, the assessment of progression is dependent on clinical, radiological, and/or biochemical criteria that are subject to measurement error.
PFS is generally a good predictor of OS for cases where survival post-progression (SPP) is short, that is, when the post-progression noise compromising OS is not allowed to dominate; however, when OS is long relative to PFS, interpretation of their relationship is challenging.
From a regulatory standpoint, the US Food and Drug Administration and the European Medicines Agency have accepted PFS as a surrogate endpoint in many tumors and clinical scenarios, but also recommend OS as a secondary endpoint in cases where PFS is the primary endpoint.Citation1 For most new cancer treatments, an increase in OS needs to be either statistically shown or assessed as probable through demonstration of trends in data.Citation7–Citation9 This contrasts with what is commonly requested when a pharmacoeconomic analysis is needed for drug reimbursement. Pharmacoeconomic analyses examine the incremental cost per unit of efficacy, with OS as the typical unit for efficacy, to obtain an overall incremental cost per life year gained between two treatments. Therefore, challenges in obtaining meaningful OS differences are important in the reimbursement setting.
Treatment of metastatic renal cell carcinoma (mRCC) has been revolutionized in recent yearsCitation10 by the introduction of new agents such as sunitinib, sorafenib, bevacizumab, everolimus, temsirolimus, axitinib, and pazopanib.Citation11–Citation17 All except for temsirolimus were approved based on demonstrated PFS benefit alone, and authors cited the impact of subsequent treatment and crossover to explain lack of a significant OS increase.Citation10 Temsirolimus did demonstrate an OS benefit over interferon in the initial Phase III trial; however; the SPP, and also OS, was relatively short in comparison with other trials, likely due to the poor prognosis of the mRCC patients enrolled.
Broglio et alCitation18 assumed a lack of treatment effect beyond progression and showed through simulation that even a substantial benefit in PFS may be attenuated or lost as SPP increases; this is due to the random variability introduced by patient heterogeneity and subsequent therapy. They conclude that a lack of statistical significance in OS does not imply lack of improvement. Heng et alCitation10 used the Broglio et al methodology to validate the PFS-OS correlation among 1,158 mRCC patients, and concluded that the SPP diluting effect is even larger when applying the methodology to patient-level data rather than a theoretical distribution.
The AXIS (axitinib versus sorafenib in advanced metastatic renal cell carcinoma) trialCitation17 was the first to compare two TKI VEGF-R blocking drugs. The objective of the present analysis was to apply the simulation method reported by Broglio et alCitation18 to clinical trial data from the AXIS trial, in order to enable an examination of the likelihood of an OS benefit when a PFS benefit exists and SPP is long in this setting.
Materials and methods
Following Broglio et al,Citation18 OS was expressed as the sum of PFS and SPP where the progression event may be death, in which case SPP is zero. A simulated trial was considered, where the number of patients, censoring pattern, and treatment allocations would equal those in AXIS. PFS and SPP were both simulated with exponential distributions, with PFS distribution parameters chosen to be equal to median PFS in the AXIS trial (6.8 and 4.7 months for the axitinib and sorafenib arms, respectively). The SPP distribution parameter was varied, but always assumed equal for both arms. Since we assume a treatment benefit in PFS but no effect on SPP, the improvement in PFS is directly reflected in OS improvement. Censoring times were bootstrapped from the trial data to further mimic the trial situation.
For each scenario, we simulated 10,000 trials, varying median SPP in order to investigate the impact of length of SPP on the ability to detect an OS benefit. The axitinib and sorafenib treatments were compared with respect to OS in terms of the hazard ratio (HR) estimated from a Cox proportional hazard model with treatment effect as the predictor. The probability of statistical significance of PFS and OS and the estimated probability of observing HR ≥0.9 were derived and summarized. The probability of statistical significance was calculated as the fraction of simulations where a one-sided log-rank test had a P-value ≤0.025.
Simulations were conducted in the overall population and in the major subgroup of sunitinib-refractory patients. This is an important subgroup since it represents 53.8% of the total sample, and sunitinib is the standard of care in first line.
Results
Median PFS, OS, and SPP as observed in the AXIS trial is shown in . The OS difference among treatments is small relative to the difference in PFS. Thirteen simulation scenarios were run for the overall patient group; the first with median SPP equal to the pooled value from the trial (see ) and the following with increasing median SPP. Scenario data including median PFS, median SPP, the probability of statistical significance of PFS and OS, the probability of observing an OS HR ≥0.9, and the correlation between HRs for PFS and OS are shown in . The probability of statistical significance of OS is decreased from 54.7% to 6.1% when the median SPP is increased from one to 20 months. The probability of OS HR ≥0.9 ranges from 24.3% to 72.6%, and generally increases with increasing median SPP up to about 10 months, and then stabilizes. The correlation between the HRs for PFS and OS ranges from 0.11 to 0.62 and weakens when median SPP increases. The change in OS HR with increasing median SPP illustrates how the latter can increase the variability which dilutes the OS benefits.
Table 1 PFS, OS, and SPP as observed in the AXIS trial
Table 2 OS HR simulation summary
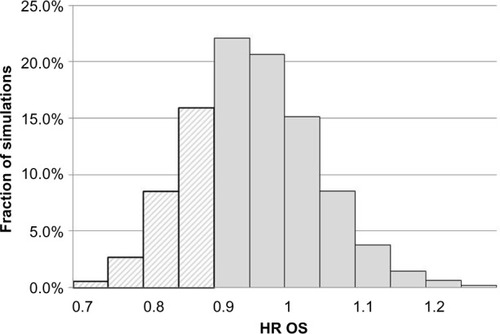
The distribution of OS HR among simulations for scenario 1 (median SPP 12.9 months based on pooled estimate from AXIS trial) is presented graphically in . In 42.7% of all simulation, the OS HR was between 0.9 and 1.0, while the corresponding fraction for OS HR below 0.9 was only 27.5%. In 29.7% of cases, the OS HR was ≥1.0 despite the large difference in median PFS between the two treatments.
Figure 1 Distribution of HRs for OS simulation 1 (mSPP is 12.9 months and the probability of statistical significance for PFS is 91.4%). Dashed bars represent simulations yielding a statistically significant OS difference (in total 27.5% of cases).
Abbreviations: HR, hazard ratio; OS, overall survival; mSPP, median survival post-progression; PFS, progression-free survival.
The probability of statistical significance of PFS and OS and the probability of observing an OS HR ≥0.9 for the sunitinib-refractory subgroup is shown in . The OS statistical significance probability is decreased from 44.8% to 4.9% when median SPP is increased from one to 20 months. The probability of OS HR ≥0.9 ranges from 32.4% to 77.7%, and similar to the overall study population, generally increases with increasing median SPP up to about 10 months.
Table 3 OS HR simulation summary for sunitinib-refractory patients
Discussion
This study builds on the simulation method devised by Broglio et alCitation18 by utilizing patient-level data from a Phase III clinical trial in mRCC, thus allowing for more realistic simulation of censoring and enrolment patterns. Our study illustrates the challenges of demonstrating an OS difference when a PFS difference exists, when making the assumption of no treatment effect on SPP. Although the HR for PFS was 0.69, the HR for OS was ≥0.9 in between 24.3% and 72.6% of the 10,000 simulations when the median SPP was varied between one and 20 months. This is because the time period until progression (during which PFS is measured) is small in relation to the median SPP. As a consequence, the probability of a statistically significant difference of OS decreased when the median SPP increased. The magnitude of change was however small when SPP was greater than 8 months and appeared to plateau when SPP reached 16 months as the censoring distribution begins to dominate the data beyond that point. This phenomenon was not present in the analysis by Broglio et al, which was performed on entirely simulated data. It is due to the fact that in the present analysis censoring time was simulated to mimic the AXIS trial to reflect the follow-up pattern in the real world situation.
A similar trend was shown for the sunitinib-refractory patient subset. These results support the conclusions regarding the impact of SPP on diluting the OS benefit not only in the overall treatment group but in the subgroup as well.
The main observation in the presented study is that increasing median SPP increases measurement noise to a level where an underlying OS difference becomes increasingly difficult to detect. This observation is not novel;Citation18,Citation19 however, it has not been previously shown using patient-level data such as demonstrated here. Our treatment arms are well balanced with respect to post-trial treatments, suggesting that the dilution is primarily due to random variation, with the impact increasing with increasing median SPP.
The use of OS as a primary endpoint for evaluating new systemic oncologic therapies is not unproblematic and is also widely debated.Citation1–Citation4,Citation20–Citation22 Therapy-specific data are diluted by imbalances in use of downstream treatments; usually it is not possible, even in clinical trials, to control the post-progression treatment use and effect, which may modify the SPP and finally the OS. The situation is sometimes accentuated in trials if crossover from standard of care treatment to the investigated drug is allowed. Taking that into account, when new active treatments become successively available, as is the case for mRCC, it will be very difficult to demonstrate any OS gain for one specific treatment even if this gain does exist.
Comparisons between modern day cancer treatments will in many cases result in a high absolute OS and a moderate OS incremental difference and therefore demand long follow-up and large trial populations. This will in turn increase trial costs and delay introduction of effective therapies into clinical practice.
PFS is the most commonly used intermediate endpoint to OS. PFS is not affected by post-progression therapy, and is accessible earlier. PFS can, however, be compromised by measurement timing yielding an overestimation and may, at least compared with OS, be biased by subjective assessments. Independent assessment of scans by radiologists blinded to treatment has been used to overcome some of these difficulties.
Whether PFS can be accepted as a primary endpoint is, indeed, often considered to depend on its value as a surrogate for OS. Surrogacy can be validated,Citation23,Citation24 and this has been done for certain tumor types, such as colorectal cancer.Citation25 Delea et al presented their mRCC results,Citation26 indicating that the treatment effect on PFS is strongly associated with the treatment effect on OS, but as Broglio et alCitation18 have shown, this correlation tends to weaken as the median SPP increases.
However, as discussed in a recent review by Fallowfield et al,Citation21 PFS can be relevant on its own, if accompanied by evidence of discernible clinical benefit for the patient. In the AXIS trial, patient-reported outcomes results based on the Functional Assessment of Cancer Therapy-Kidney Symptom Index (FKSI)-15 and FKSI Disease-Related Symptoms questionnaires demonstrated a worsening of mRCC symptoms at the end of treatment, where patients were coming off treatment mainly due to progression,Citation27 and hence delaying progression would be of value to patients.
This study was based on the primary assumption of no treatment effect on SPP. As can be seen in , median SPPs by treatment arms (overall and in the sunitinib-refractory subset) differ, although the estimates of SPP might be impacted by censoring in OS. Study limitations also include the use of an exponential distribution for PFS and SPP, selected for simplicity of use and interpretation, and without factoring in randomization stratification, which might impact the magnitude of the correlation between the HRs for PFS and OS, while having minimal impact on other OS results.
The AXIS trial compared patients in second-line, where PFS and OS would be expected to be shorter compared with a first-line setting. It may therefore be reasonable to assume that our findings are conservative and generally applicable for tumors where survival and median SPP is fairly long. Other mRCC treatments where SPP is long, such as sunitinib, sorafenib, and pazopanib, could be impacted similarly, depending on factors such as crossover, time of study conduct, and whether or not there are multiple subsequent treatments available.
Under current guidelines from regulatory agencies, new drugs can be granted market authorization in the absence of a proven effect on OS if trials have demonstrated a substantial PFS benefit and SPP is long.Citation28 Based on our simulations, a median SPP ≥10 months resulted in a probability of achieving statistical significance based on OS of less than 10% and the correlation between the PFS HR and the OS HR was less than 0.20. Reimbursement agencies and other payer bodies often in addition require evidence of cost-effectiveness. This entails quantifying the mean survival-adjusted or quality-adjusted survival gain with the new therapy, and the relationship between the additional cost and the additional benefit of the new therapy compared with standard of care, expressed as an incremental cost-effectiveness ratio.Citation29 Since it is seldom possible to derive all this information from a single clinical trial, many agencies will accept the use of economic models to combine data from several sources to produce estimates of cost-effectiveness. Even when a statistically significant benefit on OS has been demonstrated, modeling is usually needed to predict the tail of the survival curves in order to estimate the mean gain in life expectancy.Citation30 If trials have demonstrated PFS benefits but evidence on OS gains is lacking, models can be employed to predict the OS benefit from available data. Some of the factors that make it difficult to detect OS benefits in a clinical trial will also impact the ability to model OS gains from PFS; long SPP and the availability of downstream therapies will confound the link between OS and PFS, and increase the uncertainty of model estimates.
Coverage decisions made at the time of introduction of a new therapy are based on incomplete information, in particular since there is limited experience with the use of the therapy in clinical practice. Therefore, some countries have introduced “coverage with evidence development” or “risk-sharing” schemes, through which the new therapy is initially funded contingent on the accumulation of additional evidence on its effectiveness and cost-effectiveness in actual practice.Citation31 Prospective observational studies conducted after launch can provide important information on actual use and outcomes with the therapy in clinical practice. Data from administrative registries and databases can provide population-wide coverage and large sample sizes, and in some instances enable analysis of the impact of the introduction of new therapy.Citation32 Lastly, the use of PFS as the efficacy endpoint in the cost-effectiveness evaluation may be another consideration.
This paper illustrates the methodological problems with demonstrating OS gains through randomized clinical trials in patients with long SPP. If trial-demonstrated OS benefits are mandated for new drugs, the availability of new treatments for patients with long SPP will be severely limited. Considerations with the use of PFS and OS as primary or supportive endpoints need to be weighed in the context of the specific tumor being studied and the implication, not only for regulatory approval but reimbursement as well.
Disclosure
This study was sponsored by Pfizer Inc. YT and PB are employed by Pfizer Global Research, La Jolla, CA, USA, and own stock in Pfizer Inc. ÖÅ is employed by OptumInsight, Stockholm, Sweden. LJ at the time of study was employed by OptumInsight. Both ÖÅ and LJ were paid consultants to Pfizer in connection with the development of this manuscript. SN is employed by the University of Lyon, Lyon, France, has served as a consultant or adviser for Novartis and Pfizer, has received honoraria from Novartis, Pfizer, GlaxoSmithKline, and Roche, and has received research funding from Novartis, Pfizer, and GlaxoSmithKline. CC is employed by and owns stock in Pfizer Inc., New York, NY, USA.
References
- LebwohlDKayABergWBaladiJFZhengJProgression-free survival: gaining on overall survival as a gold standard and accelerating drug developmentCancer J200915538639419826358
- HotteSJBjarnasonGAHengDYProgression-free survival as a clinical trial endpoint in advanced renal cell carcinomaCurr Oncol201118Suppl 2S11S1921969807
- Di LeoABuyseMBleibergHIs overall survival a realistic primary end point in advanced colorectal cancer studies? A critical assessment based on four clinical trials comparing fluorouracil plus leucovorin with the same treatment combined either with oxaliplatin or with CPT-11Ann Oncol200415454554915033657
- SaadEDKatzAHoffPMBuyseMProgression-free survival as surrogate and as true end point: insights from the breast and colorectal cancer literatureAnn Oncol201021171219901012
- SoriaJCMassardCLe ChevalierTShould progression-free survival be the primary measure of efficacy for advanced NSCLC therapy?Ann Oncol201021122324233220497965
- SargentDJHayesDFAssessing the measure of a new drug: is survival the only thing that matters?J Clin Oncol200826121922192318421044
- EscudierBKatajaVRenal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-upAnn Oncol201021Suppl 5v137v13920555064
- National Comprehensive Cancer NetworkNCCN Clinical Practice Guidelines in Oncology Kidney Cancer Version 22010 Available from: http://www.nccn.org/professionals/physician_gls/PDF/kidney.pdfAccessed August 8, 2014
- LjungbergBCowanNCHanburyDCEAU guidelines on renal cell carcinoma: the 2010 updateEur Urol201058339840620633979
- HengDYXieWBjarnasonGAProgression-free survival as a predictor of overall survival in metastatic renal cell carcinoma treated with contemporary targeted therapyCancer2011117122637264221656741
- MotzerRJHutsonTETomczakPSunitinib versus interferon alfa in metastatic renal-cell carcinomaN Engl J Med2007356211512417215529
- EscudierBBellmuntJNegrierSPhase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survivalJ Clin Oncol201028132144215020368553
- RiniBIHalabiSRosenbergJEBevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206J Clin Oncol200826335422542818936475
- MotzerRJEscudierBOudardSEfficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trialLancet2008372963744945618653228
- SternbergCNDavisIDMardiakJPazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trialJ Clin Oncol20102861061106820100962
- HudesGCarducciMTomczakPTemsirolimus, interferon alfa, or both for advanced renal-cell carcinomaN Engl J Med2007356222271228117538086
- RiniBIEscudierBTomczakPComparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trialLancet201137898071931193922056247
- BroglioKRBerryDADetecting an overall survival benefit that is derived from progression-free survivalJ Natl Cancer Inst2009101231642164919903805
- AmirESerugaBKwongRTannockIFOcanaAPoor correlation between progression-free and overall survival in modern clinical trials: are composite endpoints the answer?Eur J Cancer201248338538822115991
- BuyseMSargentDJSaadEDSurvival is not a good outcome for randomized trials with effective subsequent therapiesJ Clin Oncol201129354719472022042965
- FallowfieldLJFleissigAThe value of progression-free survival to patients with advanced-stage cancerNat Rev Clin Oncol201291414722009075
- KornELFreidlinBAbramsJSOverall survival as the outcome for randomized clinical trials with effective subsequent therapiesJ Clin Oncol201129172439244221555691
- PrenticeRLSurrogate endpoints in clinical trials: definition and operational criteriaStat Med1989844314402727467
- BuyseMMolenberghsGCriteria for the validation of surrogate endpoints in randomized experimentsBiometrics1998543101410299840970
- BuyseMBurzykowskiTCarrollKProgression-free survival is a surrogate for survival in advanced colorectal cancerJ Clin Oncol200725335218522418024867
- DeleaTEKhuuAHengDYHaasTSoulieresDAssociation between treatment effects on disease progression end points and overall survival in clinical studies of patients with metastatic renal cell carcinomaBr J Cancer201210771059106822935581
- CellaDEscudierBRiniBPatient-reported outcomes for axitinib vs sorafenib in metastatic renal cell carcinoma: phase III (AXIS) trialBr J Cancer201310881571157823579211
- European Medicines AgencyGuideline on the evaluation of anticancer medicinal products in man Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/12/WC500119966.pdfAccessed March 2, 2012
- CartwrightWSBook Review. Methods for the economic evaluation of health care programmes, second edition. By Michael F. Drummond, Bernie O’Brien, Greg L Stoddart, George W. Torrance. Oxford: Oxford University Press, 1997J Ment Health Policy Econ1999214311967406
- DaviesABriggsASchneiderJThe ends justify the mean: outcome measures for estimating the value of new cancer therapiesHealth Outcomes Res Med201231e25e36
- EspinJRoviraJGarcaLExperiences and impact of European risk-sharing schemes focusing on oncology medicines2011 Available from: http://ec.europa.eu/enterprise/sectors/healthcare/files/docs/risksharing_oncology_012011_en.pdfAccessed August 9, 2014
- National Institute for Health and Clinical ExcellenceAxitinib for the treatment of advanced renal cell carcinoma after failure of prior systemic treatment2013 Available from: http://www.nice.org.uk/guidance/gid-tag305/resources/renal-cell-carcinoma-advanced-axitinib-final-appraisal-determination-document2Accessed August 9, 2014