
Abstract
Background
Airway hyperresponsiveness (AHR) is associated with airway inflammation and a rapid decline in lung function and is a predictor of future risk of COPD among smokers. Alveolar macrophages (AMs) from patients with COPD release a greater amount of matrix metalloproteinase (MMP)-9. We hypothesized that the imbalance between MMP-9 and tissue inhibitor of metalloproteinase-1 (TIMP-1) is related to AHR in smokers.
Patients and methods
Healthy smokers with AHR (AHR + S) or smokers without AHR (AHR − S; divided according to a methacholine challenge test) and nonsmokers without AHR (AHR − NS) were enrolled. Spirometry was performed during enrollment and repeated after 5 years. Initially, AMs recovered from bronchoalveolar lavage (BAL) fluid were cultured in the presence of p38 mitogen-activated protein kinase (MAPK) inhibitor (SB203580), MAPK kinase (MEK) 1/2 (the MEK of extracellular signal-regulated kinase [ERK] inhibitor, PD98059), or medium alone for 24 h. The release of MMP-9 and TIMP-1 in culture supernatants was measured by enzyme-linked immunosorbent assay.
Results
A greater reduction in forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC), FEV1 (as a percentage of the predicted value [%pred]), and maximal mid-expiratory flow (MMEF) was observed among AHR + S in the 5-year period. There was a higher proportion of neutrophils and a lower proportion of AMs in BAL fluid recovered from AHR + S. Compared to AMs from AHR − NS and AHR − S, AMs from nonsmokers with AHR (AHR + NS) released more MMP-9 and less TIMP-1, with an increase in MMP-9/TIMP-1 ratios. The MMP-9/TIMP-1 ratio in smokers was positively correlated with the annual decline in FEV1%pred, FVC%pred, and MMEF%pred. Both SB203580 and PD98059 significantly reduced MMP-9, but not TIMP-1, from AMs of smokers.
Conclusion
AMs of AHR + NS produce excessive MMP-9 over TIMP-1, which may be a predictor of the development of airway obstruction. Inhibition of p38 MAPK and ERK suppresses the generation of MMP-9 by AMs from smokers.
Take home message
Alveolar macrophages from smokers with airway hyperresponsiveness release a greater amount of matrix metalloproteinase (MMP-9), which can be suppressed by inhibitors of p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK), and a lesser amount of tissue inhibitor of metalloproteinase-1 (TIMP-1), thus increasing the annual decline in lung function.
Introduction
COPD is the fourth leading cause of death, affecting >300 million people globally.Citation1 The diagnosis of COPD is made in patients with respiratory symptoms, persistent airflow limitation confirmed by a postbronchodilator forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) of <70%, often associated with cigarette smoking.Citation2 Tobacco smoke inhalation activates epithelial cells and alveolar macrophages (AMs) to release chemoattractants for circulating neutrophils, monocytes, and lymphocytes into the lungs.Citation3 These cells in COPD patients release a greater amount of proteinases and growth factors upon smoking, resulting in the breakdown of lung parenchyma, mucus hypersecretion, and small airway fibrosis, namely emphysema and chronic bronchitis.Citation4–Citation7
Only a subset of cigarette smokers develop COPD, and the host factors leading to airflow limitation are under active investigation. It has been postulated that airway hyperresponsiveness (AHR) predisposes the smokers’ airways to obstruction.Citation8,Citation9 AHR is defined as an increased sensitivity of the airways to direct stimuli (eg, histamine and methacholine) and/or indirect stimuli (eg, mannitol and hypertonic saline). The Lung Health Study demonstrated that AHR was strongly associated with the degree of airway obstruction in current smokers.Citation8 Current smokers with airway obstruction displayed a greater AHR than smokers with normal lung function.Citation10 Thus, smokers with AHR (AHR + S) could be prone to the development of COPD due to chronic airway inflammation and structural alternation.
The excess of protease over antiprotease expression, such as neutrophil elastase versus α1-anti-trypsin and matrix metalloproteinases (MMPs) versus tissue inhibitors of metalloproteinases (TIMPs), has been proposed for cigarette smoke-induced chronic lung disease.Citation11 MMPs are a family of >20 zinc-dependent endopeptidases with enzymatic activities in turnover and degradation of extracellular matrices. MMP-9, also known as gelatinase B or 92 kDa gelatinase, is one of the major elastolytic enzymes produced by AMs from COPD patients but are also secreted by neutrophils, epithelial cells, mast cells, and fibroblasts.Citation3 MMP-9 proteolytically digests extracellular matrix (ECM) proteins including collagens IV, V, VII, X, and XIV, gelatin, and elastin, and activates latent pro-MMP-9 and pro-MMP-13.Citation12 Epigenetically, MMPs could be modulated by methylation of the CpG sites in promoters and chromatin remodeling with histone acetylation.Citation12 Hypomethylation of MMP-9 promotor had been observed in chondrocytes in osteoarthritis, which could be associated with the increased synthesis of the cartilage-degrading enzyme.Citation13 Sirtuin1, a histone deacetylase acting on the activator protein-1 (AP-1) response element in the promotor of MMP-9, inhibits histone 3 acetylation and reduces MMP-9 in COPD.Citation14,Citation15 The activity of MMPs is also modulated transcriptionally by phosphatase and tensin homolog (PTEN) and mitogen-activated protein kinases (MAPKs), posttranscriptionally by microRNAs, and posttranslationally by reversion-inducing cysteine-rich protein with kazal motifs and a four-member family of TIMPs.Citation11 Both active form and precursor form of MMP-9 are antagonized by TIMP-1. The amount of MMP-9 released from AMs of COPD patients and smokers was greater than that of nonsmokers, but the expression of TIMP-1 was higher in sputum in chronic bronchitis and lower in emphysematous lungs.Citation16–Citation19 It is likely that the balance between MMP-9 and TIMP-1 determines the clinical phenotypes of COPD.
MMP-9 inhibition may seem an appealing strategy to treat respiratory diseases including COPD but can hardly be achieved by inhaled corticosteroids, and the effects of MMP inhibitors, blocking antibodies, and antisense technologies have not been widely recognized in clinical practice.Citation20,Citation21 MAPKs, including p38 MAPKs, c-Jun NH2-terminal kinases (JNKs), and extracellular signal-regulated kinases (ERKs), are a family of serine/threonine protein kinases activated in response to external signals (cigarette smoke, wood smoke, colonizing bacteria, oxidative stress, cytokines, and growth factors), resulting in the secretion of many mediators, which then activate further inflammatory cascades, tissue remodeling, and aging process of the lungs.Citation22 MAPK signaling is regulated by successive phosphorylation starting with mitogen-activated protein (MAP) kinase kinase kinases; then MAP kinase kinases such as MAP2K3/6, MAP2K4, and MAPK kinase (MEK) 1/2 (MAP2K1/2); and finally p38 MAPK and ERK, modulating transcription factors such as nuclear factor-κB (NF-κB) and AP-1. Little is known about the effect of MAPK inhibitors on the release of MMP-9 and TIMP-1 from AMs.
We hypothesized that the imbalance between MMP-9 and TIMP-1 contributes to AHR, which appears to be an early feature of COPD in susceptible smokers. We compared the evolution of lung function over a 5-year period, as well as the generation of MMP-9 and TIMP-1 from AMs in nonsmokers without AHR (AHR − NS) and smokers without AHR (AHR − S) and AHR + S. We also examined whether the inhibition of p38 MAPK and ERK modulates the release of MMP-9 or TIMP-1 from AMs in smokers.
Patients and methods
Studied population
Subjects aged between 20 and 75 years were recruited from our outpatient department, Chang Gung Memorial Hospital. All enrolled subjects had no abnormal radiographic findings on their chest radiograph and had a normal pulmonary function tests and negative bronchodilator response at the initial visits. Current tobacco smokers with a smoking history of at least 20 pack-years were divided into AHR − S (n=13, smokers with a dose of methacholine causing a 20% fall in FEV1 [PC20] ≥16 mg/mL in a methacholine challenge test, described later in the “Lung function and methacholine challenge testing” section) and AHR + S (n=20, smokers with a PC20 of <16 mg/mL), and only nonsmokers who had a negative methacholine challenge test were enrolled (AHR − NS, n=24). None of the recruited subjects had upper airway infection within 6 months before enrollment, tuberculosis, asthma, bronchiectasis, and systemic lupus erythematosus or took antibiotics, inhaled or systemic corticosteroids, immunosuppressants, or regular medications for extrapulmonary diseases before entering in to the study. Thirty-one subjects received fiberoptic bronchoscopy, including 10 AHR − NS, 13 AHR − S, and 8 AHR + S. All patients provided written informed consent, and the study was approved by the Ethics Committee of Chang Gung Hospital (92-099).
Lung function and methacholine challenge testing
Measurement of lung function at the beginning and a follow-up visit of ~6 years later for each enrolled participant was performed by a Spiroanalyzer ST-350R (Fukuda Sangyo Co., Ltd, Tokyo, Japan). The best of three reproducible values was chosen. A normal lung function was defined by an FEV1/FVC of ≥75%, an FEV1 of ≥80% of the predicted value (%pred), an FVC of ≥80%pred, and a maximal mid-expiratory flow (MMEF; also known as forced expiratory flow at 25%–75%) of ≥60%pred in the absence of a significant rise in FEV1 (12% and 200 mL) after bronchodilators. The rate of decline in FEV1, FVC, and MMEF was computed by dividing the total change in the 5-year follow-up period by 5, which represented an average annual rate of change.Citation23 Methacholine challenge test was performed 1 week before bronchoscopy.Citation24 The FEV1 was measured 5 min after the inhalation of phosphate-buffered saline or methacholine solution at incremental concentrations via nebulizer until the FEV1 had fallen by >20% of the starting FEV1. The log dose–response curve for methacholine was constructed as the percentage changes in FEV1 from the baseline (postbuffer) value, and the PC20 was measured by linear interpolation. AHR was defined as a PC20 of <16 mg/mL.
Bronchoalveolar lavage (BAL) and preparation of BAL cells
Lower respiratory cells were collected by BAL using fiberoptic bronchosocopy.Citation25 After premedication with intravenous atropine (0.6 mg) and midazolam (5–10 mg) and topically applied laryngeal lidocaine, a fiberoptic bronchoscope was passed through the nasal passages into the trachea under oxygen supplementation. BAL was performed using six aliquots (50 mL each) of sterile and warm 0.9% saline solution into the right fourth or fifth subsegmental bronchus. Retrieved fluid was pooled, filtered through two layers of sterile gauze, and centrifuged at 600× g for 20 min at 4°C. The cell pellet was washed sequentially and resuspended in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 5% heat-inactivated fetal calf serum (FCS; Flow Laboratories, Paisley, Scotland, UK) at 106 cells/mL. The cell viability was determined by trypan blue exclusion. The differential cell counts were performed by counting 500 cells on cytocentrifuge preparations using the modified Wright–Giemsa stain.
Cells in BAL fluid were plated in six-well Petri dishes at a concentration of 106 cells/mL for 24 h at 37°C, 5% CO2. The medium was replaced by 1 mL of fresh complete medium after culture for 24 h, and the adherent AM was incubated for a further 24 h in the presence of medium alone, medium + SB20358 (10 μM, a p38 MAPK inhibitor), or medium + PD98059 (30 μM, an MEK inhibitor). Supernatants were stored at −20°C until experimental assay.
Enzyme-linked immunosorbent assays for measurement of MMP-9 and TIMP-1 released by AMs in the culture supernatants
MMP-9 and TIMP-1 secreted by AMs in supernatants were assayed using commercially available quantitative sandwich-type enzyme-linked immunoassay kits (Amersham Life Sciences, Arlington Heights, IL, USA). Briefly, monoclonal primary antibodies were coated onto a microtiter plate, washed with PBS/Tween and blocked with PBS/10% FCS (200 μL). Standards and culture supernatant samples were then added, followed by horseradish peroxide-conjugated secondary antibodies, 5,5′-tetramethyl benzidine and hydrogen peroxide. Termination of the reaction was done by sulfuric acid (1.0 M). The color change was read spectrophotometrically at a wavelength of 450 mM, and the quantification of MMP-9 and TIMP-1 was performed by comparing the optical density of the sample with the standard curve.
Statistical analysis
Results are reported as mean ± standard error of mean. The Chi-square test was used to determine whether there was a significant difference between the expected frequencies and the observed frequencies in more categories. Differences among AHR − NS, AHR − S, and AHR + S were estimated by Kruskal–Wallis one-way ANOVA test, followed by Dunn’s post hoc multiple comparisons. The difference in smoking history between AHR − S and AHR + S was determined by Mann–Whitney test. The difference between categorical variables (sex and smoking habit) was determined by Chi-square test. To evaluate the effect of p38 MAPK and ERK inhibition, results were analyzed using the Friedman one-way ANOVA test, followed by Dunn’s post hoc multiple comparisons to the determine the differences between the control group and each treatment group. Correlations were determined by Spearman’s rank correlation. A P-value of <0.05 was accepted as statistically significant.
Results
More rapid development in airflow limitation in AHR + S
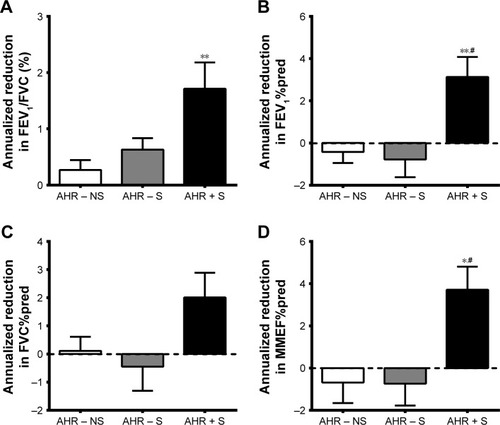
As shown in , the history of tobacco smoking during initial enrollment and the numbers of participants remaining smoking during the 5-year follow-up visit between AHR − S and AHR + S were not significantly different. The baseline pulmonary function test results were within normal ranges, but AHR + S had lower FEV1/FVC and FEV1%pred compared to AHR − NS and lower MMEF%pred compared to AHR − NS and AHR − S. AHR + S had a greater annual decline in a 5-year period from baseline in FEV1/FVC, FEV1%pred, and MMEF%pred (). The FEV1%pred and MMEF%pred of AHR + S dropped below normal, while there was no significant change of lung function in AHR − NS and AHR − S, indicating a correlation between AHR and the development of airflow limitation. The smoking history (pack-years) was not correlated with the rate of annual decline in FEV1/FVC (r=0.031, P=0.866), FEV1%pred (r=0.131, P=0.514), FVC%pred (r=0.213, P=0.235), and MMEF%pred (r=−0.045, P=0.863).
Table 1 Patient demographics and lung function
Figure 1 More rapid decline in airflow in AHR + S.
Abbreviations: AHR, airway hyperresponsiveness; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; MMEF, maximal mid-expiratory flow; %pred, as a percentage of the predicted value.
Higher percentage of neutrophils in cells in BAL fluid recovered from AHR + S
summarizes total and differential cell counts in BAL fluid, with a significant increase in cellularity recovered from BAL fluid of AHR − S compared to that of AHR − NS and AHR + S. The concentration of neutrophils was higher in AHR + S compared to those of AHR − S and AHR − NS. The number of AMs in 1 mL of BAL fluid was higher from AHR − S than from nonsmoker and AHR + S.
Table 2 Bronchoalveolar lavage fluid findings from nonsmokers and smokers
Higher MMP-9 and lower TIMP-1 productions by AMs from AHR + S
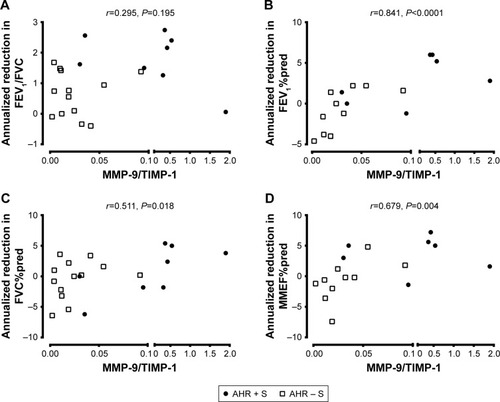
The level of MMP-9 in culture supernatant released from AMs was higher in AHR + S (10.0±4.0 ng/mL, n=8, P<0.05) than in AHR − S (1.0±0.3 ng/mL, n=13) and AHR – NS (0.7±0.2 ng/mL, n=10) (). There was a lower level of TIMP-1 released by AMs in AHR + S (39.0±10.0 ng/mL, P<0.05) than in AHR − NS (73.1±11.7 ng/mL) (). AHR + S had a significant increase in the molar ratio of MMP-9 to TIMP-1 (0.46±0.22, P<0.05) released from AM compared to AHR − S (0.025±0.007) and AHR – NS (0.006±0.002) (). Moreover, the MMP-9/TIMP-1 ratio in smokers was positively correlated with the annual decline in FEV1%pred, FVC%pred, and MMEF%pred, suggesting that the imbalance between MMP-9 and TIMP-1 is related to airflow obstruction ().
Figure 2 Greater release of MMP-9 (A) but lesser release of TIMP-1 (B) by cultured AMs from AHR + S.
Notes: AMs from AHR−NS (n=10), AHR−S (n=13) or AHR+S (n=8) were incubated for 24 hr. The level of MMP-9 (A) and TIMP-1 (B) in the culture supernatants was determined by enzyme-linked immunosorbent assays. The molar ratio between MMP-9 and TIMP-1 was also calculated (C). Values are shown as individual data point, mean, and standard error of mean. *P<0.05 compared to AHR − NS. AHR − NS, nonsmokers without AHR; AHR − S, smokers without AHR; AHR + S, smokers with AHR.
Abbreviations: AHR, airway hyperresponsiveness; AMs, alveolar macrophages; MMP-9, matrix metalloproteinase-9; TIMP-1, tissue inhibitor of metalloproteinase-1.
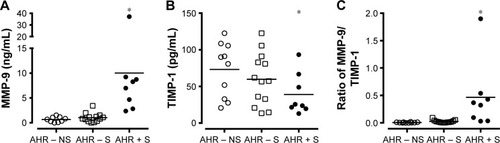
Figure 3 Association between MMP-9/TIMP-1 ratio and the development of airflow limitation.
Abbreviations: AHR, airway hyperresponsiveness; AMs, alveolar macrophages; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; MMEF, maximal mid-expiratory flow; MMP-9, matrix metalloproteinase-9; %pred, percentage of the predicted value; TIMP-1, tissue inhibitor of metalloproteinase-1.
Inhibition of p38 MAPK and ERK reduced the release of MMP-9, but not TIMP-1, by smokers’ AMs
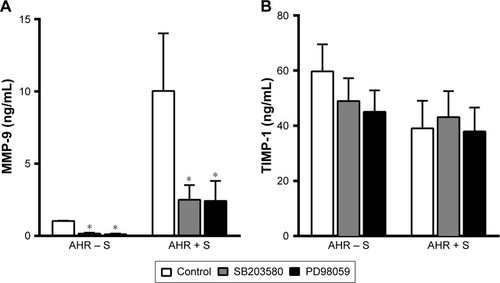
We further investigated whether the modulation of MAP kinase activities alters the release of MMP-9 and TIMP-1 by AMs from smokers. Both p38 MAPK inhibitor SB203580 (10 μM) and MEK inhibitor PD98059 (30 μM) reduced the generation of MMP-9 (2.5±1.0 ng/mL, n=8, P<0.05, and 2.4±1.4 ng/mL, n=8, P<0.05, respectively) compared to untreated controls (10.0±4.0 ng/mL, n=8) by AMs of AHR − S and AHR + S (). In contrast, neither SB203580 nor PD98059 had a significant effect on the production of TIMP-1 by AMs in AHR − S and AHR + S ().
Figure 4 Inhibition of p38 MAPK and ERK reduced the release of MMP-9, but not TIMP-1, from AMs from smokers.
Abbreviations: AHR, airway hyperresponsiveness; AMs, alveolar macrophages; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase; MMP-9, matrix metalloproteinase-9; TIMP-1, tissue inhibitor of metalloproteinase-1.
Discussion
In this study, the AHR + S had a more significant decline in FEV1/FVC, FEV1, and MMEF over a 5-year period compared to those AHR − S and AHR − NS. AMs from AHR + S released a greater amount of MMP-9 and a lesser amount of TIMP-1 than AMs from AHR − NS and AHR − S, with a higher molar ratio of MMP-9 to TIMP-1. A higher MMP-9/TIMP-1 ratio in AMs was also related to a greater loss of FEV1, FVC, and MMEF in the 5-year period. We also showed that the inhibition of p38 MAPK and ERK signaling pathways was associated with a decrease in MMP-9 production from smokers’ AMs. The results suggested that excessive MMP-9 over TIMP-1 in smokers could be related to AHR, a risk factor of COPD, and might be modulated by MAPK inhibitors.
In agreement with previously reported outcomes, accelerated deterioration of pulmonary function prior to clinical symptoms developed in AHR + S in our study. Tobacco smoking decreases FEV1/FVC, FEV1, and MMEF, indicating airway obstruction and small airway disease in smokers.Citation26 However, the incidence rate of COPD in current smokers is only 19.7/1,000 person-years (95% CI 18.1–21.4), ~2.4 times higher than in former smokers and 4.8 times higher than in never smokers.Citation27 AHR is a fundamental characteristic in asthma and also present in COPD patients and in 12%–22% of healthy subjects.Citation28 Compared to those without AHR, subjects with AHR are more likely to experience respiratory symptoms such as cough, phlegm, dyspnea, persistent wheeze, asthmatic attacks, and progressive airflow obstruction, and COPD patients with AHR suffer from a higher rate of FEV1 decline, more severe air trapping, and a double increased risk of respiratory mortality.Citation29–Citation32 We therefore suggest that AHR may be a risk factor for airway obstruction in smokers. A longitudinal study of AHR − S and AHR + S over a longer period of time will clarify whether AHR leads to a postbronchodilator FEV1/FVC of <70% in smoking population.
Increased numbers of neutrophils and macrophages in respiratory samples in parallel with exaggerated AHR had been observed in both asthmatic smokers and tobacco smoke-treated cats.Citation33,Citation34 Altered behavior of AMs is a key feature of COPD.Citation3 Increased numbers of AMs had been observed in lung parenchyma, BAL fluid, and sputum in COPD.Citation3 AMs produce fibrogenic transforming growth factor (TGF)-β, the most potent inducer of ECM generation, contributing to small airway fibrosis in COPD patients. Cigarette smoke stimulates the generation and release of neutrophils from bone marrow and prolongs the survival of neutrophils in the respiratory organs, possibly mediated by macrophages’ release of mediators, such as granulocyte–macrophage colony-stimulating factors, CXC ligand (CXCL) 8/interleukin (IL)-8, CXCL1/growth-regulated oncogene-α, leukotriene B4, and proteinases including MMP-9.Citation3 We showed an increased absolute number, although not a higher percentage, of AM in BAL fluid from smokers, and AHR + S had more neutrophils than AHR − S and AHR − NS. Recruitment of these inflammatory cells could have potentiated the inflammation and remodeling in the respiratory tracts of these smokers.
Few biomarkers are known to be positively associated with the severity of AHR in COPD apart from adiponectin and eosinophil count.Citation29,Citation35 In the present study, we highlighted the correlation between MMP-9 and AHR in smokers. We uncovered a higher ratio of MMP-9 to TIMP-1 in AMs from AHR + S, which was correlated with a greater reduction in lung function, providing a potential biomarker to predict the development of COPD in smokers. Although it is generally accepted that the excess of MMP-9’s proteolytic activity can lead to lung destruction in emphysema, there is little information linking the pathogenesis of AHR to the impact of MMP-9–TIMP-1 imbalance. AHR could be secondary to increased smooth muscle mass and contractility, infiltration of inflammatory cells, increased airway wall thickening and reduced airway caliber, infiltration of inflammatory cells, and increased production in proinflammatory and fibrogenic mediators.Citation36,Citation37 Increased numbers of AMs, neutrophils, and eosinophils in BAL fluid and elevated mRNA expression of TGF-β and MMP-9 were associated with ozone-induced AHR in a mouse model of emphysema.Citation38 There are at least three potential explanations. First, the mechanical stress elicits an increased production of ECM proteins and MMP-9 relative to TIMP-1.Citation39 Methacholine- or allergen-induced bronchoconstriction increases collagen deposition in sub-epithelial layer in parallel with the upregulation of TGF-β, stimulating the expression of MMP-9 through the activation of ERK1,2, Ras-related C3 botulinum toxin substrate 1–reactive oxygen species–NF-κB pathway, and TGF-β-activated kinase 1–NF-κB pathway, and the mechanical loads applied to the airway structural cells of individuals with AHR might be enhanced.Citation40,Citation41 Second, the disproportionate rise in MMP-9 production worsens airway remodeling and inflammation, which prompts the progression of AHR. It had been reported that asthmatic patients had remarkable expression in MMP-9 (and TIMP-1 to a lesser extent) and more prominent airway remodeling, ie, deposition of collagens and tenascin in the basement membrane, causing airflow obstruction and AHR, although conflicting results showed the other way round in the sputum specimens.Citation42,Citation43 Apart from alveolar wall matrices, MMP-9 has many other substrates such as TGF-β and CXCL8/IL-8. MMP-9 releases latent TGF-β from ECM and then is stimulated by TGF-β in a reciprocal manner, enhancing collagen deposition and small airway fibrosis. A strong correlation has been shown between the reduced airway caliber and AHR in nonasthmatic subjects with COPD, suggesting that AHR may be related to the heightened resistance secondary to geometric change of the airways.Citation36 MMP-9 facilitates chemotaxis of neutrophils by cleaving CXCL8/IL-8 into a more active form, which then amplifies the release of MMP-9 through a positive feedback loop, proteolyzes the basement membrane, and releases the chemotactic fragments from ECM to recruit inflammatory cells, compatible with the higher proportion of neutrophils in the BAL fluid from AHR + S and high MMP-9 in the present study.Citation44 Third, some AHR + S may be asthmatic, although all subjects enrolled in our study had negative bronchodilator test results. Patients with asthma exacerbation have an elevated MMP-9 level in their blood, sputum, and BAL fluid, and the ratio of MMP-9 to TIMP-1 was higher in BAL fluid from children with symptomatic asthma.Citation45 It is conceivable that more MMP-9 and less TIMP-1 released by AMs lead to further inflammatory process and remodeling and is associated with AHR and the subsequent lung function decline in smokers.
We also provided evidence that pharmacological inhibition of p38 MAPK and ERK suppresses the release of MMP-9, but not TIMP-1, from smokers’ AMs in vitro. Cigarette smoking induces marked activation of p38 MAPK and ERK.Citation46 AMs from patients with corticosteroid-insensitive severe asthma and COPD have increased expression of phosphorylated p38 MAPK.Citation47,Citation48 Reduced MMP-9 production has been observed after the inhibition of p38 MAPK, JNK, and ERK using epigallocatechin-3-gallate in phorbol 12-myristate 13-acetate-induced macrophages.Citation49 Cigarette-induced emphysema in Wistar rats is associated with the phosphorylation of p38 MAPK and ERK (c-Jun NH2-terminal kinase) and the activation of NF-κB signaling, leading to an increase in MMP-9 together with a reduction in TIMP-1, while oral erythromycin administration prevented emphysema and inflammation by reducing MMP-9 via the inhibition of p38 MAPK, ERK, and NF-κB.Citation50 Simvastatin attenuates cigarette smoke extract-induced activation of ERK, NF-κB, and AP-1, and upregulation of MMP-9 in rat AMs.Citation51 Inhibition of MAPKs, especially p38 MAPK, appears to be a logical approach to eliminate airway inflammation. Several clinical trials for inhalational p38 MAPK inhibitors, such as RV568 (now known as JNK-49095397), PF-30715455, and AZD-7624, are currently ongoing.Citation52 Clarithromycin and rosiglitazone also block the cigarette smoke extract-induced MMP-9 production in animal models.Citation53,Citation54 Modification of MMP-9 expression by either MAPK inhibitors or approved agents, which modulate MMP-9 expression, may be an additional therapeutic strategy to ameliorate the deterioration of lung function in AHR + S.
Conclusion
Our finding supports the hypothesis that AMs from AHR + S produce an excess of MMP-9 to TIMP-1, accompanied by a more severe deterioration of airway obstruction in these subjects, indicating a new predictor to identify smokers vulnerable to COPD. We also confirm the observation that the generation of MMP-9 can be hampered by the inhibition of p38 MAPK and ERK, which may be a potential therapeutic target to prevent the development of AHR and progressive airflow limitation in smokers.
Acknowledgments
This work was supported by the National Science Research Project (NMRP) grant 92-2314-B-182A-178 – and Chang Gung Memorial Hospital Research Project Grant CMRPG3B1323.
Disclosure
The authors report no conflicts of interest in this work.
References
- Global Burden of Disease Study CGlobal, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013Lancet2015386999574380026063472
- VogelmeierCFCrinerGJMartinezFJGlobal strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD Executive SummaryAm J Respir Crit Care Med2017195555758228128970
- BarnesPJInflammatory mechanisms in patients with chronic obstructive pulmonary diseaseJ Allergy Clin Immunol20161381162727373322
- ChungKFAdcockIMMultifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destructionEur Respir J20083161334135618515558
- BchirSNasrHBBouchetSConcomitant elevations of MMP-9, NGAL, proMMP-9/NGAL and neutrophil elastase in serum of smokers with chronic obstructive pulmonary diseaseJ Cell Mol Med20172171280129128004483
- de BoerWIHauCMvan SchadewijkAStolkJvan KriekenJHHiemstraPSExpression of epidermal growth factors and their receptors in the bronchial epithelium of subjects with chronic obstructive pulmonary diseaseAm J Clin Pathol2006125218419216393673
- LeeSHLeeSHKimCHIncreased expression of vascular endothelial growth factor and hypoxia inducible factor-1alpha in lung tissue of patients with chronic bronchitisClin Biochem2014477–855255924463065
- TashkinDPAltoseMDBleeckerERThe lung health study: airway responsiveness to inhaled methacholine in smokers with mild to moderate airflow limitation. The Lung Health Study Research GroupAm Rev Respir Dis19921452 pt 13013101736734
- FrewAJKennedySMChan-YeungMMethacholine responsiveness, smoking, and atopy as risk factors for accelerated FEV1 decline in male working populationsAm Rev Respir Dis199214648788831416413
- Garcia-GarciaJMHernandezJRMartinez-MunizMAAirways reactivity, atopy and bronchoalveolar lavage in male smokers with airflow obstructionRespiration19966341992048815965
- NavratilovaZKolekVPetrekMMatrix metalloproteinases and their inhibitors in chronic obstructive pulmonary diseaseArch Immunol Ther Exp (Warsz)2016643177193
- LoffekSSchillingOFranzkeCWSeries “matrix metalloproteinases in lung health and disease”: biological role of matrix metalloproteinases: a critical balanceEur Respir J201138119120821177845
- RoachHIYamadaNCheungKSAssociation between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regionsArthritis Rheum200552103110312416200590
- NakamaruYVuppusettyCWadaHA protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9FASEB J20092392810281919376817
- GaoZYeJInhibition of transcriptional activity of c-JUN by SIRT1Biochem Biophys Res Commun2008376479379618823944
- OhnishiKTakagiMKurokawaYSatomiSKonttinenYTMatrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysemaLab Invest1998789107710879759652
- VignolaAMRiccobonoLMirabellaASputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitisAm J Respir Crit Care Med19981586194519509847290
- RussellRECulpittSVDeMatosCRelease and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol200226560260911970913
- LimSRocheNOliverBGMattosWBarnesPJChungKFBalance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10Am J Respir Crit Care Med20001624 Pt 11355136011029344
- VandenbrouckeREDejonckheereELibertCA therapeutic role for matrix metalloproteinase inhibitors in lung diseases?Eur Respir J20113851200121421659416
- CulpittSVMaziakWLoukidisSNightingaleJAMatthewsJLBarnesPJEffect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med19991605 Pt 11635163910556133
- BarnesPJKinases as novel therapeutic targets in asthma and chronic obstructive pulmonary diseasePharmacol Rev201668378881527363440
- WangXZhangHXSunBXCross-shift airway responses and long-term decline in FEV1 in cotton textile workersAm J Respir Crit Care Med2008177331632017975204
- PopaVATS guidelines for methacholine and exercise challenge testingAm J Respir Crit Care Med20011631292293
- WangCHLiuCYLinHCYuCTChungKFKuoHPIncreased exhaled nitric oxide in active pulmonary tuberculosis due to inducible NO synthase upregulation in alveolar macrophagesEur Respir J19981148098159623681
- KupermanASRikerJBThe variable effect of smoking on pulmonary functionChest19736356556604703618
- TerzikhanNVerhammeKMHofmanAStrickerBHBrusselleGGLahousseLPrevalence and incidence of COPD in smokers and non-smokers: the Rotterdam StudyEur J Epidemiol201631878579226946425
- LeiYGaoYChenJGLCCI1 rs37973: a potential genetic predictor of therapeutic response to inhaled corticosteroids in Chinese chronic obstructive pulmonary disease patientsSci Rep201774255228186150
- TkacovaRDaiDLVonkJMAirway hyperresponsiveness in chronic obstructive pulmonary disease: a marker of asthma-chronic obstructive pulmonary disease overlap syndrome?J Allergy Clin Immunol2016138615711579.e1027345171
- XuXRijckenBSchoutenJPWeissSTAirways responsiveness and development and remission of chronic respiratory symptoms in adultsLancet19973509089143114349371166
- O’ConnorGTSparrowDWeissSTA prospective longitudinal study of methacholine airway responsiveness as a predictor of pulmonary-function decline: the Normative Aging StudyAm J Respir Crit Care Med1995152187927599868
- WalkerPPHadcroftJCostelloRWCalverleyPMLung function changes following methacholine inhalation in COPDRespir Med2009103453554119081234
- KolahianSShahbazfarAATayefi-NasrabadiHTiotropium effects on airway inflammatory events in the cat as an animal model for acute cigarette smoke-induced lung inflammationExp Lung Res201440627228724784973
- ShimodaTObaseYKishikawaRIwanagaTInfluence of cigarette smoking on airway inflammation and inhaled corticosteroid treatment in patients with asthmaAllergy Asthma Proc20163745058
- PostmaDSRabeKFThe asthma-COPD overlap syndromeN Engl J Med2015373131241124926398072
- CockcroftDWDavisBEMechanisms of airway hyperresponsivenessJ Allergy Clin Immunol2006118355155916950269
- AtkinsonJJLuteyBASuzukiYThe role of matrix metalloproteinase-9 in cigarette smoke-induced emphysemaAm J Respir Crit Care Med2011183787688421057003
- LiFWiegmanCSeiffertJMEffects of N-acetylcysteine in ozone-induced chronic obstructive pulmonary disease modelPLoS One2013811e8078224260479
- SwartzMATschumperlinDJKammRDDrazenJMMechanical stress is communicated between different cell types to elicit matrix remodelingProc Natl Acad Sci U S A200198116180618511353845
- KrsticJSantibanezJFTransforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cellsScientific World Journal2014201452175424578639
- GraingeCLLauLCWardJAEffect of bronchoconstriction on airway remodeling in asthmaN Engl J Med2011364212006201521612469
- MatsumotoHNiimiATakemuraMRelationship of airway wall thickening to an imbalance between matrix metalloproteinase-9 and its inhibitor in asthmaThorax200560427728115790981
- HoshinoMNakamuraYSimJShimojoJIsogaiSBronchial subepithelial fibrosis and expression of matrix metalloproteinase-9 in asthmatic airway inflammationJ Allergy Clin Immunol199810257837889819295
- OverbeekSABraberSKoelinkPJCigarette smoke-induced collagen destruction; key to chronic neutrophilic airway inflammation?PLoS One201381e5561223383243
- GrzelaKLitwiniukMZagorskaWGrzelaTAirway remodeling in chronic obstructive pulmonary disease and asthma: the role of matrix metalloproteinase-9Arch Immunol Ther Exp (Warsz)20166414755
- KochAGiembyczMStirlingRGEffect of smoking on MAP kinase-induced modulation of IL-8 in human alveolar macrophagesEur Respir J200423680581215218990
- BhavsarPHewMKhorasaniNRelative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthmaThorax200863978479018492738
- RendaTBaraldoSPelaiaGIncreased activation of p38 MAPK in COPDEur Respir J2008311626917959643
- WangQMWangHLiYFInhibition of EMMPRIN and MMP-9 expression by epigallocatechin-3-gallate through 67-kDa laminin receptor in PMA-induced macrophagesCell Physiol Biochem20163962308231927832636
- ZhouXGuDHouGErythromycin attenuates metalloprotease/anti-metalloprotease imbalance in cigarette smoke-induced emphysema in rats via the mitogen-activated protein kinase/nuclear factor-kappaB activation pathwayMol Med Rep20171552983299028358431
- KimSEThanh ThuyTTLeeJHSimvastatin inhibits induction of matrix metalloproteinase-9 in rat alveolar macrophages exposed to cigarette smoke extractExp Mol Med200941427728719299917
- NormanPInvestigational p38 inhibitors for the treatment of chronic obstructive pulmonary diseaseExpert Opin Investig Drugs2015243383392
- HouGYinYHanDWangQYKangJRosiglitazone attenuates the metalloprotease/anti-metalloprotease imbalance in emphysema induced by cigarette smoke: involvement of extracellular signal-regulated kinase and NFkappaB signalingInt J Chron Obstruct Pulmon Dis20151071572425897215
- NakamuraMWadaHHondaKClarithromycin ameliorates pulmonary inflammation induced by short term cigarette smoke exposure in micePulm Pharmacol Ther201535606626363279