
Abstract
Introduction
COPD is a chronic inflammatory disease of lung. The inflammatory response in COPD is associated with neutrophils, macrophages, T lymphocytes, and bronchial epithelial cells, and occurs mainly in the small airway, leading to irreversible airflow limitation.
Methods
In order to investigate the microRNA–mRNA interaction in the microenvironment of the COPD airway, we used next-generation sequencing and bioinformatics in this study.
Results
We identified four genes with microRNA–mRNA interactions involved in COPD small-airway bronchial epithelial cells: NT5E, SDK1, TNS1, and PCDH7. Furthermore, miR6511a-5p–NT5E interaction was found to be involved in small-airway bronchial epithelial cells, large-airway bronchial epithelial cells, and alveolar macrophages.
Conclusion
Our results showed that miR6511a-5p–NT5E interaction plays an important role in COPD, which might be associated with cell–cell contact, activation of leukocytes, activation of T lymphocytes, and cellular homeostasis. These findings provide new information for further investigations of the COPD microenvironment, and may help to develop new diagnostic or therapeutic strategies targeting the bronchial epithelium for COPD.
Introduction
COPD affected 174.5 million people and caused 3.2 million deaths worldwide in 2015.Citation1 It is progressive and usually causes major disability.Citation2 COPD is a chronic inflammatory disease of the lung. The inflammatory response of COPD is associated with inhaled noxious substances, such as smoke and air pollution.Citation3 Neutrophils, macrophages, and T lymphocytes are increased in the lung with COPD.Citation3 As a defensive barrier of airways, bronchial epithelial cells are also affected in the inflammatory process of COPD.Citation4 After being stimulated by inhaled insults, bronchial epithelial cells initiate immune and inflammatory responses, including recruiting macrophages, neutrophils, and dendritic cells.Citation4 The inflammatory response occurs mainly in the small airway and results in significant airflow limitation, which is not fully reversible. With the progression of COPD, the inflamed small airways undergo remodeling processes involving airway-wall thickening, reduced airway diameter, and increasing resistance to respiratory flow.Citation5 Recently, epithelial–mesenchymal transition caused by repeated stimulation was identified as a potential source of fibroblasts and myofibroblasts that could contribute to the chronic remodeling of airways.Citation6
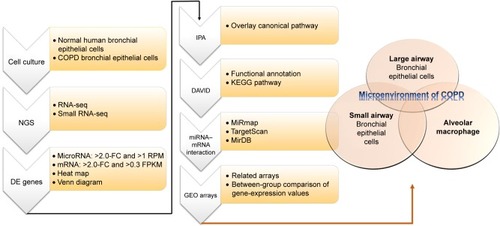
MicroRNAs have been recognized as important regulators in the gene regulation of COPD.Citation7 Downregulated miR128 in smokers, downregulated miR181d and miR30a-3p in COPD lung tissue, and increased miR223 in lungs of smokers have been demonstrated.Citation7 In a study investigating muscle wasting of COPD patients, the results showed that increased miR424-5p was associated with muscle wasting.Citation8 These studies showed that microRNA–mRNA interactions may play important roles in COPD, as we have demonstrated in asthma.Citation9 In order to investigate microRNA–mRNA interactions in the microenvironment of COPD airways, we used COPD bronchial epithelial (DHBE) cells and normal bronchial epithelial cells to perform next-generation sequencing (NGS) and bioinformatic analyses ().
Figure 1 Flowchart of study design.
Abbreviations: DE, differentially expressed; FC, fold change; RPM, reads per million; FPKM, fragments per kilobase of transcript per million mapped reads; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Methods
Primary cells
Primary normal human bronchial epithelial (NHBE) and DHBE cells were purchased from Lonza (Basel, Switzerland). Cell cultures were performed according to the manufacturer’s protocol. Cells were grown in bronchial epithelial cell basal media (BEGM™ Bronchial Epithelial Cell Growth Medium BulletKit™; Lonza), supplemented with 2 mL bovine pituitary extract, 0.5 mL hydrocortisone, 0.5 mL human epidermal growth factor (hEGF), 0.5 mL epinephrine, 0.5 mL transferrin, 0.5 mL insulin, 0.5 mL retinoic acid, 0.5 mL triiodothyronine, and 0.5 mL GA1000. Cells were maintained at 37°C in a 5% CO2 incubator and passaged with Reagent-Pack (Lonza) containing trypsin–EDTA (EDTA, trypsin neutralizing solution, and HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid]-buffered solution). Cells were harvested for NGS analysis after cultivation from primary cells for one generation.
Next-generation sequencing
Expression profiles of microRNAs and mRNAs were examined using NGS. Total RNA of both normal and DHBE cells was extracted using Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Purified RNA was then quantified at OD260 using an ND-1000 spectrophotometer (NanoDrop; Thermo Fisher Scientific) and quality assessed with a 2,100 bioanalyzer with RNA 6000 LabChip kit (Agilent Technologies, Santa Clara, CA, USA) at Welgene Biotech (Taipei, Taiwan).
For small-RNA library construction and deep sequencing, samples were prepared using an Illumina sample-preparation kit according to the TruSeq small-RNA library sample-preparation guide. In summary, total RNA was ligated with 3′ and 5′ adaptors and reverse-transcribed into cDNA followed by polymerase chain-reaction amplification. The harvested cDNA constructs were fractionated by size and purified with 6% polyacrylamide-gel electrophoresis, and bands containing 18–40-nucleotide RNA fragments (140–155 nucleotides in length with both adapters) were selected. Libraries were then sequenced on an Illumina instrument (75SE cycle, single-end) and sequencing results processed with Illumina software. For small-RNA-sequence analysis, sequencing data were applied to go through a filtering process to obtain qualified reads. Trimmomatic was used to trim or remove reads according to quality scores.Citation10 Qualifying reads were then analyzed using miRDeep2 to clip the 3′ adapter sequence and shorter reads (<18 nucleotides) removed, before aligning reads with the human genome from the University of California, Santa Cruz.Citation11 Because microRNAs are usually mapped to several genomic locations, only reads mapped perfectly to the genome five or more times were used for microRNA detection. MiRDeep2 was used to estimate expression levels of microRNAs. The criteria for microRNA selection were fold change >2, and reads per million >1.
For transcriptome sequencing, a library was constructed with Agilent’s SureSelect Strand specific RNA-library-preparation kit for 75SE (single-end or paired-end) and sequencing performed on the Solexa platform. Sequences were determined directly using sequencing-by-synthesis technology via the TruSeq SBS kit. Raw sequences were obtained from Illumina Pipeline software bcl2fastq version 2.0 and expected to generate 30 million reads per sample. The sequences generated went through a filtering process to obtain qualifying reads. Trimmomatic was implemented to trim or remove the reads according to the quality score.Citation10 Qualifying reads were analyzed using TopHat/Cufflinks to estimate gene-expression level, calculated as fragments per kilobase of transcript per million mapped reads.Citation12 For differential-expression analysis, the Cummerbund statistical package was employed to perform statistical analyses of gene-expression profiles. The reference genome and gene annotations were retrieved from the Ensembl database.
MiRmap database analysis
MiRmap is an open-source software library providing comprehensive microRNA-target prediction (http://mirmap.ezlab.org).Citation13 Putative target genes can be identified by calculating the complementary ability of microRNA–mRNA interactions. The predictor also estimates mRNA-repression strength for ranking potential candidate targets by a combination of thermodynamic, evolutionary, probabilistic, and sequence-based features. The prediction results provide a list of putative target genes with MiRmap score, which is a predictive reference value. In this study, the criterion for selection of putative microRNA targets was MiRmap score ≥99.
MirDB database analysis
MirDB is an online database for predicting microRNA-target and functional annotations. In MirDB, MirTarget was used for predicting all the targets. MirTarget was designed by analyzing microRNA-target interactions from high-throughput sequencing experiments. MirDB can predict microRNA targets in five species, including human, mouse, rat, dog, and chicken.Citation14,Citation15
TargetScan database analysis
TargetScan is an online database for predicting biological targets of microRNAs. It searches for the presence of conserved 8mer, 7mer and 6mer sites, matching the seed region of each microRNA. Predictions are ranked based on the predicted efficacy of targeting or by their probability of conserved targeting.Citation16
DAVID database analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID), which integrates multiple functional annotation databases, including Gene Ontology, biological processes, or Kyoto Encyclopedia of Genes and Genomes pathways, is a powerful tool for gene-function classification (https://david.ncifcrf.gov).Citation17 A list of interesting genes can be classified into clusters of related biological functions, signaling pathways, or diseases by calculating the similarity of global annotation profiles with an agglomeration algorithm. It also provides an Ease score, which is a modified Fisher’s exact P-value. The reference score represents how specifically the user genes are involved in the category (eg, signaling pathways). In this study, we selected an Ease score of 0.1 as default and 1 to extend clustering range.
Gene Expression Omnibus database analysis
The Gene Expression Omnibus (GEO) is a web database that collects submitted high-throughput gene-expression data of microarrays, chips, or NGS (https://www.ncbi.nlm.nih.gov/geo).Citation18 Microarrays of accession numbers GSE4498 (small-airway bronchial epithelial cells), GSE5056 (large-airway bronchial epithelial cells), and GSE2125 (alveolar macrophages) were used in this study. GSE4498 assessed gene expression (HG133 Plus 2.0 array) in ten phenotypically normal smokers compared to 12 matched nonsmokers.Citation19 GSE5056 assessed gene expression in 13 phenotypically normal smokers and nine normal nonsmokers.Citation20 GSE2125 assessed gene expression in 15 cigarette smokers and 15 nonsmokers.Citation21 Raw data extracted from GEO were replotted and statistically analyzed with Student’s t-test using GraphPad Prism 7 software (GraphPad Software, La Jolla, CA, USA).
Ingenuity Pathway Analysis
Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA, USA) contains a large database with detailed and structured findings reviewed by experts. This IPA database was derived from thousands of biological, chemical, and medical studies, and provides researchers with quick searching. IPA also enables analysis, integration, and recognition of data from gene and single-nucleotide-polymorphism arrays, RNA and small-RNA sequencing, proteomics, and many other biological experiments. In addition, deeper understanding and identification of related signaling pathways, upstream regulators, molecular interactions, disease processes, and candidate biomarkers are also available in IPA.Citation22
Results
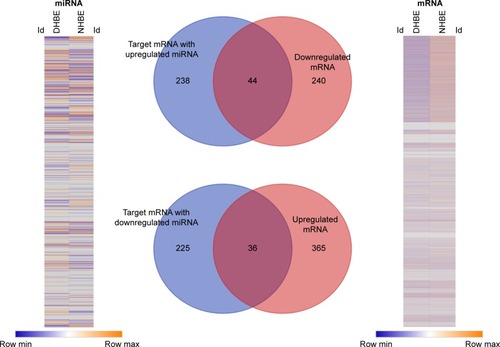
In order to identify the potential microRNA–mRNA interactions involved in the homeostasis of COPD epithelium, we analyzed the expressions of genes and microRNAs using NGS data from DHBE and NHBE cells (). The gene-expression heat map of differentially expressed genes revealed 685 genes with fold change >2, including 284 downregulated genes and 401 upregulated genes. The heat map of microRNA expression revealed 144 microRNAs with fold change >2. Based on the MiRmap web-based database, we predicted 543 mRNA as targets of these 144 microRNAs, including 282 targets of upregulated microRNAs and 261 targets of downregulated microRNAs. Venn diagrams of microRNA–mRNA interactions showed that 44 genes were downregulated and 36 genes upregulated in DHBE cells compared to NHBE cells. These 80 dysregulated genes with potential microRNA–mRNA interactions are listed in .
Table 1 Dysregulated genes with potential microRNA–mRNA interactions in COPD bronchial epithelial cells
Figure 2 Identification of genes with potential microRNA–mRNA interactions in COPD bronchial epithelial cells.
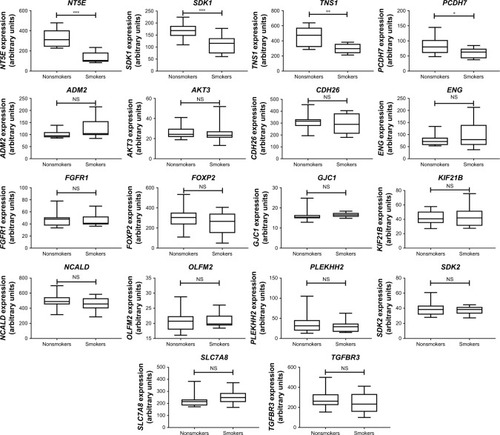
We then used IPA to analyze the function of these 80 dys-regulated genes. Possible pathways related to these 80 genes are demonstrated in . shows pathways related to 44 downregulated genes, and shows pathways related to 36 upregulated genes. In , epithelial adherens junction signaling, T-helper (TH)-1-and TH2-activation pathways, and the TH2 pathway were involved in downregulation of the 44 genes. This implies that some of these genes may be associated with the function of bronchial epithelium adherence and immunity of the COPD lung. Because most microRNAs could have down-regulated gene expression, we focused on the interaction between upregulated microRNA and downregulated mRNA. To analyze the possible mechanisms of these 44 down-regulated genes in DHBE cells, we used DAVID (). The results showed that these 44 genes were involved in the functions of membrane, transmembrane helix, transmembrane, nucleotide bonding, cell adhesion, calcium, disulfide bonds, and signals. We further analyzed the disease and function of 44 downregulated genes with IPA and then performed meta-analyses with MirDB and TargetScan. The meta-analyses showed 17 upregulated microRNA with interactions with 18 downregulated mRNAs (). To validate the identified 18 downregulated mRNAs in clinical COPD samples, we used the GEO database and selected a representative microarray (accession number GSE4498) that contains bronchial epithelial cells in small airways from 12 nonsmokers and ten smokers. Four genes with significant differences in mRNA expression between nonsmokers and smokers were found in GSE4498: NT5E, SDK1, TNS1, and PCDH7 ().
Table 2 Downregulated genes with microRNA–mRNA interactions and their functions
Figure 3 Functional analysis of dysregulated genes identified in COPD epithelial cells by Ingenuity Pathway Analysis (IPA).
Notes: The 80 identified dysregulated genes with potential microRNA–mRNA interactions were analyzed by IPA. (A) Pathways related to 44 downregulated genes; (B) pathways related to 36 upregulated genes.
Abbreviations: TR, thyroid hormone receptor; RXR, retinoid X receptor; PTEN, phosphatase and tensin homolog; FAK, focal adhesion kinase; NFAT, nuclear factor of activated T-cell.
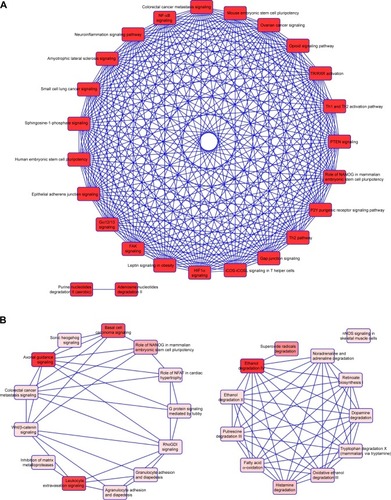
Figure 4 Possible mechanisms of dysregulated genes identified in COPD epithelial cells analyzed by Database for Annotation, Visualization, and Integrated Discovery (DAVID).
Notes: Functional annotation of the 44 downregulated genes was determined by gene ontology using DAVID. These 44 genes are involved in the functioning of membrane, transmembrane helix, transmembrane, nucleotide bonding, cell adhesion, calcium, disulfide bonds, and signals.
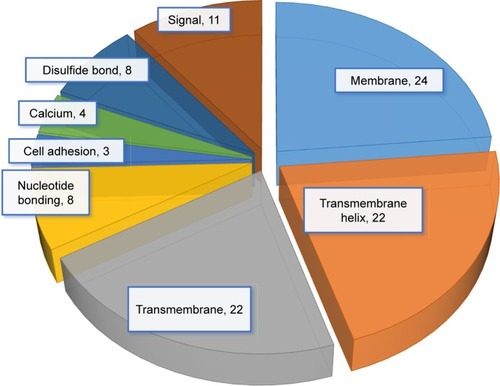
Figure 5 Gene Expression Omnibus (GEO) database analysis of 18 downregulated genes with potential microRNA–mRNA interactions in COPD small airway.
Abbreviation: NS, not significant.
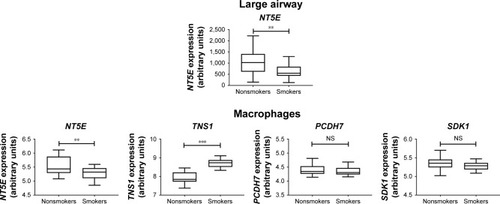
To investigate further the role of these four genes that were downregulated in the small airway of COPD, in the microenvironment of other parts of the COPD airways, we used the GSE5056 (large-airway bronchial epithelial cells) and GSE2125 (alveolar macrophages) databases for meta-analyses. The results showed NT5E was the only downregulated gene in bronchial epithelial cells of the small airway, large airway, and alveolar macrophages (). Expression data for TNS11, PCDH, and SDK1 were not available in the GSE5056 data set. Our data showed that hsa-miR6511a-5p was the possible upstream regulator for NT5E (). NT5E may be involved in cell–cell contact, activation of leukocytes, activation of T lymphocytes, and cellular homeostasis. We also analyzed the 36 upregulated mRNAs and found only two interactions: miR20b-5p–SLC46A3 and miR145-5p–FLI1. However, of SLC16A3 and FLI1 overexpression was not validated in the data sets of GSE4498, GSE5056, or GSE2125 (data not shown).
Figure 6 Gene Expression Omnibus database analysis of the four genes downregulated in COPD small airway, large airway, and alveolar macrophages.
Abbreviation: NS, not significant.
Discussion
COPD is a disease with abnormal inflammatory airway processes and can lead to structural changes in lung parenchyma, airways, and vessels.Citation23 Macrophages in the lung are important immunoeffector cells. Lung macrophages provide innate and adaptive immunoresponses to inhaled foreign matters in the lung.Citation24 Bronchial epithelial cells of large and small airways and alveolar macrophages could be considered important components of the COPD microenvironment. Further investigation of gene regulations in this COPD microenvironment may provide potential therapeutic implications and knowledge of COPD pathogenesis. In this study, we analyzed the gene-expression profiles of microRNAs and mRNAs in NHBE and DHBE cells by NGS. We used bioinformatics to investigate the potential molecular mechanisms of gene regulation. Using MiRmap for predicting targets and Venn diagrams for intersection analysis, we focused on 44 potential microRNA–mRNA interactions (upregulated microRNA–downregulated mRNA). After meta-analysis with MirDB and TargetScan website, we focused on 17 upregulated microRNAs with interactions with 18 downregulated mRNAs. We further analyzed these 18 genes in the GEO database. The database we chose was GSE4498 (microarray data from small-airway bronchial epithelial cells), and four genes with significant and identical changes were found. In order to analyze the role of these four genes in the microenvironment of COPD, we further analyzed these four genes in the GSE5056 (large-airway bronchial epithelial cells) and GSE2125 (alveolar macrophages) databases. The results showed that NT5E was the only significantly downregulated gene. According to our NGS data, miR6511a-5p was the upstream regulator of NT5E. In the IPA analysis, NT5E was associated with cell–cell contact, activation of leukocytes, activation of T lymphocytes, and cellular homeostasis.
The NT5E gene is associated with cellular functions dependent on G-protein-coupled receptors specific for adenosine. These functions include proliferation, apoptosis, and activation.Citation25 In a study of glioma cell lines, inhibition of ecto-5′-nucleotidase, the enzyme encoded by the NT5E gene, led to significant reduction in glioma-cell proliferation.Citation26 In a study of human airways, ecto-5′-nucleotidase was found to be responsible for the production of adenosine on the mucosal surface of human airway epithelial cells and was an important factor in the regulation of adenosine-mediated epithelial functions.Citation27 In a radiation-induced pulmonary fibrosis study, inhibition of ecto-5′-nucleotidase significantly reduced radiation-induced lung fibrosis.Citation28 In our study, the downregulation of NT5E discovered from NGS analysis was validated in the GEO databases of COPD small airway, large airway, and alveolar macrophages. Accompanying the results of IPA analysis, NT5E, regulated by miR6511a-5p, could be an important factor in the microenvironment of the COPD airway.
In our study, downregulation of SDK1, TNS1, and PCDH1 discovered from NGS analysis was also validated in the GEO database of COPD small-airway epithelial cells. The SDK1 gene encodes SDK1, which is an adhesion molecule. SDK1 is activated by cellular stress, such as with reactive oxygen species.Citation29 SDK1 may be also associated with asbestos-exposure-related lung malignancies.Citation29 In a study investigating cross-species cancer genes, SDK1 was both significantly amplified and significantly deleted, which may indicate that the SDK1 gene resides in unstable regions of the genome.Citation30 Therefore, the down-regulated SDK1 shown in our study might have been related to tumorigenesis and associated with lung cancer in COPD patients.
The TNS1 gene encodes tensin 1, which is involved in fibrillar adhesion formation. It might also be related to cell migration and cartilage development.Citation31 A genome-wide association study of COPD also showed that patient forced expiratory volume in 1 second (FEV1) or FEV1:forced vital capacity (FVC) ratio was associated with common variants at locus 2q35 in TNS1.Citation32 A study on TNS1 function showed significant reduction in proliferation and migration in endothelial cells isolated from TNS1-knockout mice or those silenced with TNS1 siRNA.Citation33 In our study, we found an opposite direction for TNS1 expression in small-airway bronchial epithelial cells and alveolar macrophages of COPD lungs. These findings suggest that proliferation and migration might be decreased in DHBE cells and increased in COPD alveolar macrophages.
The PCDH7 gene encodes Pcdh7, which is a member of the cadherin superfamily.Citation34 The PCDH7 gene product is an integral membrane protein involves in cell–cell recognition and adhesion.Citation35 A study of gastric cancer demonstrated that Pcdh7 inhibited cell migration and invasion.Citation35 Therefore, downregulation of PCDH7 could promote cell migration. In the nervous system, protocadherins are widely expressed transmembrane proteins, and overexpression of Pcdh7 would cause intrinsic apoptotic pathways in primary cortical neurons.Citation36 Our IPA analysis also showed that PCDH7 was associated with neurological signs. However, in the pathogenesis of COPD, Pcdh7 might be more related to migration and adhesion of bronchial epithelial cells of the small airway.
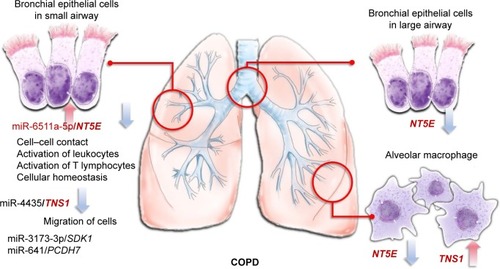
To date, there has been neither cure against COPD nor effective biomarkers for diagnosing COPD. In this study, we identified potential microRNA–mRNA interactions that regulate the homeostasis of DHBE cells. Our results showed that the miR6511a-5p–NT5E interaction plays an important role in COPD and might be associated with cell–cell contact, activation of leukocytes, activation of T lymphocytes, and cellular homeostasis. We also found that miR3173-3p–SDK1, miR4435–TNS1, and miR641–PCDH7 interactions might be associated with COPD pathogenesis in small-airway bronchial epithelial cells (). However, there are several limitations in this study. Firstly, it is not easy to obtain bronchial epithelial cells from either healthy subjects or COPD patients. Potentially dysregulated microRNAs, mRNAs, and their interactions in COPD were generated from only one normal subject and one COPD patient. Although we used GEO to validate the differentially expressed genes and TargetScan and MirDB to validate the interaction between microRNA and gene expression predicted by MiRmap, these findings need to be validated in more clinical samples. Secondly, the DHBE cells, cultivated in the same normal epithelial culture medium, had already gone out of the actual microenvironment in COPD airways. As this was a screening and bioinformatic study, our findings need further validation.
Figure 7 MicroRNA–mRNA interactions in the microenvironment of COPD.
Conclusion
In conclusion, this study showed that miR6511a-5p–NT5E, miR3173-3p–SDK1, miR4435–TNS1, and miR641–PCDH7 interactions might play important roles in the bronchial epithelium of COPD. These findings provide new information for further investigations of the COPD microenvironment, and may help in development of new diagnostic or therapeutic strategies targeting bronchial epithelium for COPD.
Acknowledgments
The authors gratefully acknowledge the support of research grants from the Ministry of Science and Technology (MOST 106-2314-B-037-016-MY2, MOST 104-2320-B-037-014-MY3), Kaohsiung Medical University Hospital Research Foundation (KMUHS10601, KMUH106-6R15), Kaohsiung Medical University Research Foundation (105KMUOR05), and the KMU-KMUH Co-Project of Key Research (KMU-DK 107009 from Kaohsiung Medical University).
Disclosure
The authors report no conflicts of interest in this work.
References
- GBD 2015 Chronic Respiratory Disease CollaboratorsGlobal, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015Lancet Respir Med20175969170628822787
- EcenarroPSIguiñizMITejadaSPManagement of COPD in end-of-life care by Spanish pulmonologistsCOPD Epub2018320
- SindenNJStockleyRASystemic inflammation and comorbidity in COPD: a result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidenceThorax2010651093093620627907
- GaoWLiLWangYBronchial epithelial cells: the key effector cells in the pathogenesis of chronic obstructive pulmonary disease?Respirology201520572272925868842
- CosioMGSaettaMAgustiAImmunologic aspects of chronic obstructive pulmonary diseaseN Engl J Med2009360232445245419494220
- PainMBermudezOLacostePTissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotypeEur Respir Rev20142313111813024591669
- StolzenburgLRHarrisAThe role of microRNAs in chronic respiratory disease: recent insightsBiol Chem2018399321923429148977
- ConnollyMPaulRFarre-GarrosRmiR-424-5p reduces ribosomal RNA and protein synthesis in muscle wastingJ Cachexia Sarcopenia Muscle20189240041629215200
- SheuCCTsaiMJChenFWIdentification of novel genetic regulations associated with airway epithelial homeostasis using next-generation sequencing data and bioinformatics approachesOncotarget2017847826748268829137293
- BolgerAMLohseMUsadelBTrimmomatic: a flexible trimmer for Illumina sequence dataBioinformatics201430152114212024695404
- FriedländerMRMackowiakSDLiNChenWRajewskyNmiRD-eep2 accurately identifies known and hundreds of novel microRNA genes in seven animal cladesNucleic Acids Res2012401375221911355
- TrapnellCRobertsAGoffLDifferential gene and transcript expression analysis of RNA-seq experiments with TopHat and CufflinksNat Protoc20127356257822383036
- VejnarCEZdobnovEMMiRmap: comprehensive prediction of microRNA target repression strengthNucleic Acids Res20124022116731168323034802
- WongNWangXmiRDB: an online resource for microRNA target prediction and functional annotationsNucleic Acids Res201543D146D15225378301
- WangXImproving microRNA target prediction by modeling with unambiguously identified microRNA-target pairs from CLIP-ligation studiesBioinformatics20163291316132226743510
- AgarwalVBellGWNamJWBartelDPPredicting effective microRNA target sites in mammalian mRNAsElife20154e05005
- HuangDWShermanBTTanQThe DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene listsGenome Biol200789R18317784955
- CloughEBarrettTThe Gene Expression Omnibus DatabaseMethods Mol Biol201614189311027008011
- TilleyAEHarveyBGHeguyADown-regulation of the Notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2009179645746619106307
- CarolanBJHeguyAHarveyBGLeopoldPLFerrisBCrystalRGUp-regulation of expression of the ubiquitin carboxyl-terminal hydro-lase L1 gene in human airway epithelium of cigarette smokersCancer Res20066622107291074017108109
- WoodruffPGKothLLYangYHA distinctive alveolar macrophage activation state induced by cigarette smokingAm J Respir Crit Care Med2005172111383139216166618
- ThomasSBonchevDA survey of current software for network analysis in molecular biologyHum Genomics20104535336020650822
- SohalSSEpithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD)Respir Investig2017552104113
- YamasakiKEedenSFLung macrophage phenotypes and functional responses: role in the pathogenesis of COPDInt J Mol Sci2018192E58229462886
- FaustherMLavoieEGGoreeJRBaldiniGDranoffJANT5E mutations that cause human disease are associated with intracellular mistrafficking of NT5E proteinPLoS One201496e9856824887587
- BraganholETamajusukuASBernardiAWinkMRBattastiniAMEcto-5′-nucleotidase/CD73 inhibition by quercetin in the human U138MG glioma cell lineBiochim Biophys Acta2007177091352135917643826
- PicherMBurchLHHirshAJSpychalaJBoucherRCEcto 5′-nucleotidase and nonspecific alkaline phosphatase: two AMP-hydrolyzing ectoenzymes with distinct roles in human airwaysJ Biol Chem200327815134681347912560324
- WirsdörferFde LeveSCappucciniFExtracellular adenosine production by ecto-5′-nucleotidase (CD73) enhances radiation-induced lung fibrosisCancer Res201676103045305626921334
- Mäki-NevalaSSarhadiVKKnuuttilaADriver gene and novel mutations in asbestos-exposed lung adenocarcinoma and malignant mesothelioma detected by exome sequencingLung2016194112513526463840
- MattisonJKoolJUrenAGNovel candidate cancer genes identified by a large-scale cross-species comparative oncogenomics approachCancer Res201070388389520103622
- BrunnerMMillon-FrémillonAChevalierGOsteoblast mineralization requires β1 integrin/ICAP-1-dependent fibronectin depositionJ Cell Biol2011194230732221768292
- RepapiESayersIWainLVGenome-wide association study identifies five loci associated with lung functionNat Genet2010421364420010834
- ShihYPSunPWangATensin1 positively regulates RhoA activity through its interaction with DLC1Biochim Biophys Acta185320151232583265
- YoshidaKYoshitomo-NakagawaKSekiNSasakiMSuganoSCloning, expression analysis, and chromosomal localization of BH-protocadherin (PCDH7), a novel member of the cadherin superfamilyGenomics19984934584619615233
- ChenHFMaRRHeJYProtocadherin 7 inhibits cell migration and invasion through E-cadherin in gastric cancerTumour Biol2017394 1010428317697551
- XiaoHSunZWanJHouSXiongYOverexpression of protocadherin 7 inhibits neuronal survival by downregulating BIRC5 in vitroExp Cell Res20183661718029548751