
Abstract
The field of biomarker research has almost reached unmanageable proportions in chronic obstructive pulmonary disease (COPD). The developments of new technology platforms have generated a huge information data base, both cross sectionally and increasingly, longitudinally. The knowledge emerging provides an enormous potential for understanding the disease pathophysiology, for developing markers specific for long-term outcomes, and for developing new therapeutic strategies. However, the excitement must be tempered with an understanding of the limitations of the data collection techniques, and of the variations in disease state, activity, impact, and progression. Nevertheless, the most crucial aspect in interpreting the current literature is the recognition of the relatively superficial characterization of what is a complex group of pathological processes with a common end point of airflow limitation. The current review explores some of these issues together with those areas where real progress appears to have been made, and provides caution on interpretation.
Introduction
“Biomarkers” have become a hot topic in the study and treatment of chronic obstructive pulmonary disease (COPD). In simple terms, a biomarker is a measurable characteristic that reflects the presence, severity, or state of a disease. For instance, whilst spirometry reflects the presence and dyspnea the severity or impact of COPD, temperature may reflect an infectious state. Furthermore, a change in the expression of a biomarker may reflect progression, the risk of progression, or the response to treatment, whilst genes and their products reflect the likelihood of disease emergence. Thus biomarkers can be specific cells, molecules, genes, gene products, organ functions, or general clinical characteristics that reflect the damage done, or the process that the damage sets in motion or the process that leads to the damage, or can indicate the prognosis and/or response to therapy. More recently the use of general technology platforms has led to an exponential increase in information, accompanied by uncertainty as to whether this reflects cause or effect.
The current article attempts to review (at least in part) this enormous field together with some guidelines, based on potential pitfalls and on what we need to know. In general the “hot” questions we need answers to are:
Can we identify the susceptible individual before the disease state develops?
Can we identify “early” disease whilst it is still relatively asymptomatic?
Can we predict the rate of progression of the disease?
Can we identify or confirm the key pathophysiological processes, leading to new drug discovery?
Can we identify factors that predict the response to current treatment or short Phase II clinical trials that provide confidence to undertake more extensive and definitive Phase III studies?
Can we be sure of the impact of our new strategies on the patient, in the present or future?
Defining COPD status
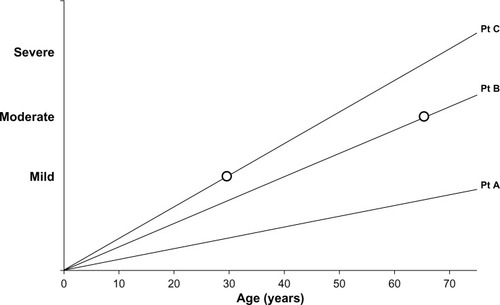
COPD is a slowly progressing disease, although the rate of progression varies. This is an important concept as it impacts upon three features of COPD, namely, the disease activity, its severity, and the impact on the patient. The activity of the disease process will influence the current or future severity in a time dependent manner, ie, a highly active disease process will produce more severe disease at an earlier age. Similarly, the severity of the disease will have a variable impact on the patient, dependent upon the rate at which it developed. Small changes that develop rapidly are more likely to have a greater impact on the younger patient than the same change developing slowly will have on a more elderly patient (see ).
Figure 1 Line graph representation of disease progression in 3 idealized patients. Patient (Pt) A has slow disease progression and with age may notice or report few symptoms (ie, low impact/low disease activity/mild disease). Patient B has greater progression and symptoms become noticeable in the 60s (moderate impact/moderate disease activity/moderate disease). Patient C has rapid disease progression leading to symptoms in the 30s (major impact/high disease activity/mild disease).
In addition, it is increasingly recognized that COPD, although defined by major changes in spirometry, has several distinct phenotypes (clinically and pathologically) as well as additional features, such as the exacerbation complex (also consisting of many pathophysiological processes) and the recognized comorbidities, which may have some common physiological yet distinct pathological mechanisms.Citation1 “Omics” is a neologism that aims at the collective characterization and quantification of pools of biological molecules that translate into the structure, function, and dynamics of an organism. Trying to understand the complexities of the enormous data generated by the “omics” platforms in COPD (proteomics, genomics, metabolomics, transcriptomics etc) is almost impossible without a similar high-quality and detailed patient characterization, and all biomarker studies need to be interpreted in this light.
Clinical biomarkers
Chest hyperinflation, low body mass index (BMI), the use of accessory muscles of respiration, and prolonged expiration have always been the physical markers of the presence of airflow obstruction, especially that occurring with a predominant emphysema phenotype. However the objective measures of lung physiology have been the “gold standard” biomarkers to date. In particular, not only has spirometry been the lynchpin of diagnosis but forced expired volume in 1 second (FEV1) has been central to our description of severity. It is a marker of disease progression and response to therapy, as well as a sound predictor of mortalityCitation2 and a weak indicator of future exacerbations.Citation3 Nevertheless, it only measures one aspect of COPD and relates poorly to the presence of emphysemaCitation4 or to patient-reported quality of life,Citation5 and its deterioration is not linear with time.Citation6
However spirometry remains central to our thinking of COPD, requires little equipment, is relatively simple to perform and interpret (with appropriate training), and hence is a useful tool to detect “early” disease (at least when sufficiently advanced to fulfill the cross-sectional diagnostic criterion). Yet, even the term “early disease” is interpreted in different ways. To a patient, this may be understood as the stage in the disease when the symptoms or their impact is mild, whereas to physiologists, it is understood to occur when the spirometry is abnormal and yet is relatively close to the normal range. This concept is summarized in , showing that the speed with which airflow obstruction develops will influence the symptoms perceived by the patient, which will clearly be dependent upon the activity of their lifestyle and expectations.
Furthermore, the presence of comorbidity and other physiological impairment (gas trapping and defects of gas transfer) will also influence the overall impact on the patient. Indeed, the impact, as determined by health status tools, is also a fair guide to long-term mortality.Citation7 However, the poor correlation of health status with physiology suggests such tools only partly measure the same thing. Mortality is related to many factors, including the degree of breathlessness,Citation8 exercise limitation,Citation9 and low BMI.Citation10 These markers predict the same end point but do not necessarily reflect the same thing. For this reason, composite scores, such as the BODE index (Body mass index, airflow Obstruction, Dyspnoea and Exercise), may be better predictors of long-term mortality.Citation11 Because these relatively insensitive tests have been studied for many years, their use in assessing patients’ prognosis, progression, and response to treatment has been well documented. However, although useful as cross-sectional measures of cohort risks, these composite markers are less robust in assessing individual patients,Citation12 and a progression of health status can occur independent of spirometry, even though progression of both also predict mortality.Citation12 However, the composite markers do fulfill two other criteria, namely, that changes, both by decline or improvement, also relate to mortality,Citation13 although may not be predictive of progression per se.
Patient-reported outcomes
Symptoms are included in many patient-reported outcomes and can be scored, in their own right, to provide both an indicator of the disease impact in individual patients but also, of progression and, importantly, the response to treatment. This alone consists of a large literature of assessment and validation. A good start for the interested reader is the validation of the COPD Assessment Test and its comparison with other patient-reported outcomes (which again highlights the weak relationship to spirometry).Citation14 Nevertheless, these subjective tests do act as a biomarker of the impact of the disease on the patient, even if this is not entirely (or even only partly) explained by the current objective measures (a disjunction between disease severity and impact).
Radiology
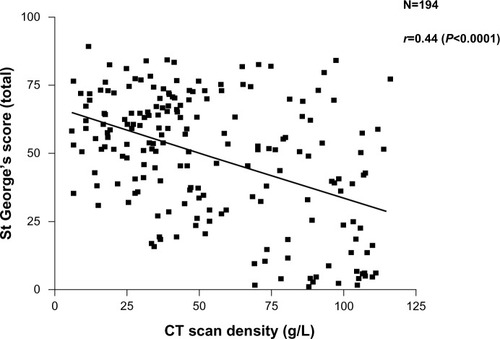
More recently, the widespread use of high-resolution computed tomography (CT) scans and other radiological techniques has been applied to COPD populations. The CT scan is now the marker of choice to identify and quantify the presence and type of emphysema. This patient phenotype also relates to health status (though also weakly) as shown in . Emphysema is a good predictor of mortality,Citation15,Citation16 can be present with normal spirometry,Citation17 and is the outcome of choice in treatments aimed at improvingCitation18 or slowing down the progressionCitation19 of emphysema. CT scanning is recognized as the most sensitive test of progression in patients with emphysema,Citation20 being significantly more sensitive than spirometry and even gas transfer, in such patients. More recently, further refinements of CT analysis have enabled the relationship between small airways disease and emphysema to be characterized in more detail,Citation21 and the preliminary data suggest that small airways disease may precede the development of radiological emphysema. The implications of this for our understanding of pathophysiology clearly requires further study.
Figure 2 Relationship between health status recorded as the SGRQ score and decreasing lung density as a marker of emphysema. Individual patient data points are shown and the significance of the correlation is given.
Radiology can also study the airways, indicating responses to bronchodilator therapyCitation22 and the presence and distribution of accompanying bronchiectasis, which is also a predictor of mortalityCitation23 (although the reasons are currently unclear).
Chronic bronchitis
The chronic bronchitis phenotype received little attention as a marker until recently and is now recognized as a predictor of disease progressionCitation24 and recurrent exacerbations.Citation25 The latter defines a further clinical phenotype (and hence marker) likely to respond to anti-inflammatory therapy, both by the inhaled routeCitation26 and oral administration.Citation27
Despite the well-tried and accepted roles of these “clinical” biomarkers, in more recent research, the term has come to mean a fluid sample test that is specific, particularly in terms of the pathophysiological processes taking place. Such research was initially based on a hypothesis-testing strategy, but the development and ready availability of technology platforms has caused this to become a data-collecting and possibly, a hypothesis-generating strategy. The latter relies on extensive data collection, which is then probed for patterns (generally the “omics” strategy) that may become informative about the potential process(es) taking place.
Biomarkers based on pathophysiology
The generally accepted concept is that the development of COPD occurs in susceptible individuals as a result of an enhanced inflammatory response to inhaled substances, like cigarette smoke. However, not only is COPD a generically defined syndrome but, inflammation itself is a highly complex, interrelated network of cells, cytokines, and other proteins that both amplify and dampen down the process – in part, this represents a balance between the destructive elements and the reparative ones. However, once damage has occurred to the delicate structures of the lung, repair cannot completely restore function, especially in the alveolar regions. This has been best demonstrated in animal models of emphysema, where a single challenge has been shown to result in the loss of lung elastin and, although it reaccumulates, the development of emphysema.Citation28 To date, the majority of biomarker studies in COPD have concentrated on the damage process.
Our understanding of the mechanisms involved in the pathophysiology of COPD relates back to the single observation that alpha-1-antitrypsin (AAT)-deficiency (AATD) resulted in an increased susceptibility to the development of early-onset basal emphysema.Citation29 The cascade of events that followed this observation resulted in many studies of AAT phenotypes in COPD, but apart from the recognition of the Pi ZZ severe form of deficiency and some very rare null genes, little was found. However, even the deficiency itself is not a highly sensitive marker since many affected patients remain healthy and with normal lung function. The true incidence of the deficiency and disease and hence, the sensitivity of even this genetic/biochemical marker remains unknown.
The mechanism involved became relatively easy to define. AAT is a specific inhibitor of serine proteinases, and elastaseCitation30 and proteinase 3,Citation31 released by activated neutrophils, have both been shown to produce emphysematous lesions in experimental animals. Since neutrophil recruitment to the lung results in the release of these enzymes, elastin digestion during transit should be excessive in the presence of low concentrations of AAT. Completion of the pathophysiological axis required a chemoattractant signal and cell activation, adhesion, and migration, as summarized in . The figure also summarizes the relevance of fluid sampling related to the parts of the process being studied.
Figure 3 The pathological process involved in emphysema. (1) cigarette smoke activates macrophages (2), epithelial cells (3) and airway neutrophils (4) to release pro inflammatory cytokines and neutrophil chemoattractants. At the same time oxidant stress in smoke damages local airway proteins (3). Harvesting airway secretions (A) detects markers of these effects including the influence of airway colonisation (5) and local mucus over production. The chemokines activate endothelial cells and circulating neutrophils (6) leading to adhesion and migration. Blood biomarkers (C) reflect these events. Migrating neutrophils ± local activated macrophages destroy connective tissue releasing specific fragments into the lymph and together with locally produced chemokines circulate into the circulation where they can be detected (B).
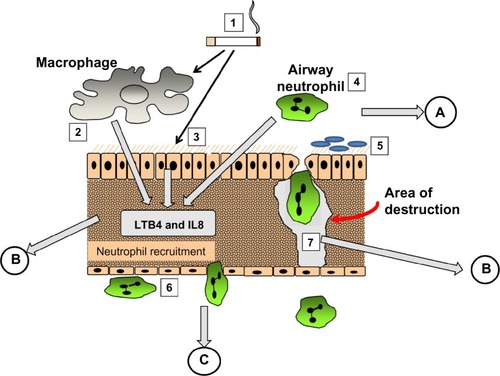
It was strongly believed that monitoring the destructive process would identify a biomarker that would be increased with progression and decreased by effective therapy. Assays were developed to detect elastin-breakdown products/peptides and the cross-linking peptides, desmosine and isodesmosine. Lung elastin is a very long-lived connective tissue,Citation32 and hence, breakdown products should not be a feature of health. Unfortunately elastin is not confined to the lung, and these elastin-breakdown products, despite being elevated in patients with neutrophilic lung diseases, do not help with the management of even AATD. There is some evidence that these elastin-breakdown products increase with disease progressionCitation33 but do not mark any therapeutic benefit.Citation34 Furthermore a variety of other inflammatory lung diseases and variable clinical states also affect the levels of those markers,Citation35 and for many reasons, their validity remains controversial. It is possible that only specific elastase-generated fragments would be useful and hence results may depend on both enzyme-cleavage preparation of antigen and monoclonal antibody.Citation36 More recently, renewed interest in the role of desmosine (an elastin cross-linking peptide) has occurred, reviewed extensively by Luisetti et alCitation34 and shown to relate to lung physiology.Citation37 The relationship with spirometry was weak but that to gas transfer was stronger, consistent with elastin breakdown being more a feature of emphysema. However, studies of elastin breakdown may be more relevant if assessed in airways secretions.Citation38 So despite the studies that evaluated lung elastin-breakdown products as far back as the 1970s and strong evidence that this is a key process in the development of emphysema, its validation as a biomarker remains an unresolved problem.Citation34
Elastase and other proteinases
The release of elastase from activated neutrophils within the lung tissues is necessary to cause the proteolytic damage implicated, at least, in emphysema. This would seem, therefore, to be a much more relevant and direct biomarker of this clinical/pathological phenotype. The problem is that although elastase release in the lung interstitium is likely to be critical for matrix degradation at this site, the enzyme is rapidly inactivated (even in AATD), especially by local inhibitors, even if already bound to elastin.Citation39 An alternative approach has been to measure the enzyme activity in the lung secretions. Sputum elastase activity is detectable in usual COPD but only during neutrophilic exacerbations,Citation40 again reflecting the capacity of the local inhibitors. However, although elastase activity is more easily detectable in AATD patients, even in the stable state,Citation41 it is also present in the secretions of bronchiectasis patients withCitation42 and withoutCitation43 cystic fibrosis. Since these latter diseases do not develop emphysema, the data suggest neutrophil recruitment more locally (bronchial circulation), sparing the interstitium, in response to bacterial load,Citation44 and overwhelming local- and serum-derived inhibitors play a role, potentially damaging the airway epithelium and local host defenses.Citation45
Indeed, other enzymes are often detected in these same secretions, including proteinase 3,Citation46 metalloproteinases,Citation47 and cysteine proteinases,Citation48 all of which have been implicated in emphysema but which also form part of an independent proteinase cascade.Citation49 Whatever the final pathway, all these enzymes and their inhibitors may become both specific and nonspecific biomarkers. Thus, whether these markers provide insight into airways disease above and beyond simple associations or merely reflect sputum purulence (see Exacerbations section) remains to be determined.
For the emphysema process, bronchoalveolar lavage (BAL) would be the only viable option for the study of markers in the airway. Although this approach has been used for more general inflammation markers (see General inflammation markers section), it has added little to the data because of the difficulty in obtaining a true alveolar sampling. However, in AATD, lavage, AAT, and elastase inhibitory capacity have been used to support the benefit of AAT augmentation therapy,Citation50 which has also been reflected in sputum studies.Citation51
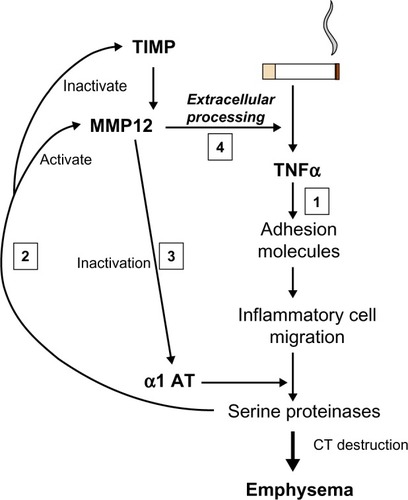
More recently, an alternative approach to the detection of lung elastase activity has been developed and at least partly validated. As enzyme is released from neutrophils, there is a finite area and time within which the enzyme remains active. Specific cleavage of the surrounding substrate can produce a biochemical footprint of enzyme activity. A specific cleavage product of fibrinogen (Aα-Val360) has been shown to relate to lung physiology, be reproducible, increase during exacerbations, and to decrease with augmentation therapy, in AATD.Citation52 Concentrations were also found to be higher than normal in usual COPD and to be related to physiological and radiological measure of emphysema as well as radiological progression of emphysema in the lower zone;Citation53 this suggests a similar elastolytic process is at least partly responsible even in the presence of normal AAT. These latter findings may well relate to the abnormal neutrophil function seen in usual COPD (see The neutrophil section). Whether studies of similar markers of other elastolytic enzymes will also be informative remains to be seen. However, of interest, it should be noted that as part of the proteinase cascade, neutrophil elastase (NE) is responsible for the activation of the cysteine proteinase cathepsin B.Citation54 Although this enzyme is not inhibited by AAT, the inhalation of AAT, which would inactivate NE, was also shown to result in the inactivation of cathepsin B,Citation55 suggesting the latter is also a downstream biomarker of local NE activity. In the same vein NE activates matrix metalloproteinases (MMPs),Citation56 and these enzymes are responsible for tumor necrosis factor (TNF)α processing,Citation57 which, in its own right, drives neutrophilic inflammation. Thus, understanding these parts of the inflammatory cascade provides, not only potential therapeutic targets but also, potential biomarker readouts of efficacy ().
Figure 4 Interrelation of proteinases and TNF. Cigarette smoking leads to TNF release and sequential events leading to emphysema (1). Serine proteinases released by recruited neutrophils activate MMP12 and inactivate its’ cognate inhibitor/s (2). MMP12 inactivates α1AT facilitating its own activation by serine proteinases (3). MMP12 then leads to extracellular processing of the interaction of TNF with its receptor (4) facilitating the main pathway (1).
The neutrophil
Since the identification of NE as a potential mediator of emphysema, studies of this cell have become central to our understanding of the end point of tissue destruction in this subtype of COPD. There is extensive literature documenting evidence of the activation of neutrophils and their adhesion molecules in COPD. However, few of these studies have determined whether this is COPD-specific or reflects lung neutrophilic disease states in general. However, airway neutrophils have been shown to relate to the decline in lung functionCitation58 and emphysema quantified by CT scan.Citation59 Nevertheless, the findings of such studies (as with all inflammatory mediators) beg the question of whether the observations are physiological, caused by the “severity” of the disease, or pathological, causing the severity of the disease itself. The data implicating the neutrophil is extensive but remains circumstantial. Although it is recognized that the degree of emphysema relates to the amount of NE in the lungCitation60 and that NE destroys elastin, for this to be pathological would require that there be an abnormal response but not in those not demonstrating the phenotype (ie, healthy age-matched smokers). AATD provides another control since the pathological mechanism (low AAT) is well established, and disease severity (to obviate any physiological response) and treatment (to overcome any influences of drugs) can be matched to patients with usual COPD.
It was recognized in 1987 that the polymorphonuclear leukocytes (PMN) from emphysema patients with COPD behaved abnormally. The cells demonstrated a rapid motility and an excessive ability to destroy connective tissue.Citation61 This was related to increased endothelial adhesion under-flowCitation62 and reflects a defective chemotactic response to routine chemoattractants.Citation63 These features are not found in healthy smokers and matched patients with AATD. The defect has been linked to the abnormal expression of phosphoinositide 3 (PI3) kinase and may reflect excessive production by phosphatidylinositol (3,4,5)-triphosphate (PIP3), a genetic defect of regulation, or defective breakdown by the phosphates, phosphatase and tensin homolog (PTEN) and SH2 domain-containing inositol 5′-phosphatase 1 (SHIP1). Thus further studies are required to determine the exact mechanism, but it does reflect a pathologically relevant trait that needs further validation, including determining whether this is a feature of susceptible smokers alone and relates to the rate of progression. Nevertheless, recent data has shown that activated neutrophils in usual COPD colocalize to the area of the lung where the emphysema develops.Citation59 The signal is also clearly different to healthy controls and AATD patients and provides a platform for early Phase II studies of neutrophil-modifying therapies.
This whole pathophysiological hypothesis of the role of the neutrophil in the development and progression of disease (at least of emphysema) has therefore a long and well-established history based on a human genetic model. With all this knowledge, the potential for biomarker discovery would seem relatively straightforward, and yet, no such validated biomarker that fulfils all the necessary requirements (marks the disease process, reflects the activity of the disease process, is stable, predicts progression, and identifies patients amenable to therapy) has emerged. With this lesson as an example, all other markers need similar understanding and background before true advances in specific disease management can occur.
The eosinophil
The eosinophil is more classically associated with asthma. However, one study in COPD has shown that an increase in the proportion of eosinophils in induced (or spontaneous) sputum is indicative of a good spirometric response to inhaled corticosteroids.Citation64 Furthermore a recent publication has also indicated that this marker (reflected in blood eosinophils) also identifies patients who are likely to benefit from oral steroid therapy for exacerbations.Citation65 Importantly, the study also suggested that the noneosinophilic group faired less well with steroid therapy. These studies would be consistent with an asthma/COPD overlap syndrome and provide guidance for a more steroid-orientated treatment strategy. Hence, eosinophils can be seen to fulfill one of the requirements for a biomarker. Although bronchodilator reversibility, per se, influences spirometric decline,Citation66 it remains unknown whether it is the eosinophilic subset that marks the susceptible, yet steroid-responsive patient or predicts future progression.
Oxidative stress
The other key process implicated in the pathophysiology of COPD is oxidative stress. Cigarette smoke itself contains high concentrations of oxygen-free radicals with the potential to damage tissues and proteins, and stimulate inflammation. Despite an efficient antioxidant defense, there has been clear and repeated evidence of oxidant stress in COPD. Again, whether this is pathological or physiological (a general response-to-smoking) remains unclear. This topic was extensively reviewed in 2008Citation67 and more recently in 2013,Citation68 and the oxidant and proteinase story has been brought together in a further recent review.Citation69 However, to date, the markers of oxidative stress have not been widely used other than to reflect the process itself. Validation of these markers and their use in supporting effective antioxidant therapy is awaited.
Genetic studies have, however, cast some light on the potential pathological, rather than physiological, nature of the ability to deal with oxidative stress (see Technology platforms section).
Inflammation markers
Although the preceding sections addressed relatively specific marker studies in COPD that were based on clinical and pathophysiological processes, the rapidly expanding literature on biomarkers has taken a more generic stance. It is widely accepted that the majority of diseases in the COPD syndrome have inflammation as the key underlying mechanism, and studies of a vast array of mediators have been undertaken, addressing three distinct questions.
Is it/are they elevated in COPD?
Does it/do they relate to the physiological markers (usually spirometric) of severity?
Does it/do they reflect a specific patient phenotype or a therapeutic target?
Using the search terms “biomarkers and COPD”, in excess of 1,000 papers can be found that have been published since 2008. It is clearly beyond the scope of the current review to discuss such a large body of literature, so selective aspects have been chosen to illustrate the general principles required for interpretation (as few have emerged to answer most of the questions outlined in the introduction). C-reactive protein (CRP) is probably the most nonspecific marker of inflammation and yet, has been widely studied in COPD. At present, over 300 articles can be identified via PubMed, using the search words of “CRP and COPD”. CRP levels, even those within the accepted normal range, predict future cardiac events in a general population,Citation70 and they are implicated in the pathophysiology of vascular disease.Citation1 There is an inverse relationship between plasma CRP levels and lung function, even in subjects assumed to be healthy.Citation71 The levels are elevated in COPD patients without evidence of ischemic heart disease; these are stable and are reduced with inhaled corticosteroids,Citation72 although this does not reflect a disease-modifying effect.
There has been no clear mechanism to implicate CRP in the pathophysiology of COPD, nor does it reflect known genetic polymorphisms, suggesting it is merely a general marker of the underlying inflammatory process associated with COPD.Citation73 However, the data did suggest that high CRP was associated with an increased odds ratio for hospitalization, although probably reflecting disease severity. The conclusions were based on a large population cohort and as such, only indicate trends and will have little impact on managing individual patients.Citation73
CRP levels rise during exacerbations particularly when there is an increased neutrophilic influx due to a bacterial cause.Citation74 In addition, a raised CRP in the stable state predicts recurrent exacerbations due either to a failure to completely resolve the first episode or a persistent underlying airway colonization that predisposes to further episodes.Citation75
Fibrinogen has also been widely studied in COPD and has been well covered in a recent review.Citation76 Fibrinogen is also an acute-phase protein and is raised in COPD (as part of the systemic effects of the disease), increases during exacerbations, and is higher in those with frequent exacerbations and those with more severe airflow obstruction. It may also play a role in comorbidities, especially vascular disease and potentially cardiovascular death due to an increased clotting tendency. Again, no genetic polymorphisms have been identified, indicating fibrinogen is also more likely to be a reflective feature of COPD. Although studies indicate that therapy can reduce fibrinogen levels, it remains unknown whether this affects progression or other outcomes. If so, fibrinogen could act as an ideal biomarker at least for some aspects of COPD, and its management and further studies are awaited. The references related to these data are included in the review article.Citation76
Many other inflammatory markers have been measured in COPD, and some seem to be persistently raised. The nonspecific nature of these changes provides little insight into management or pathophysiology. Surfactant protein D (SPD) is a lung-derived protein. SPD is elevated in the plasma in COPD and falls with corticosteroid therapy,Citation77 suggesting that steroids reduce systemic leak from the lungs as part of a general anti-inflammatory effect. However, in one study, the changes in SPD following inhaled therapy were also reflected in the degree of improvement of health status and FEV1, in the short term,Citation78 suggesting that lung inflammation itself affects these features. Of interest, the CRP and interleukin (IL)-6 did not change, suggesting that the effects were acting locally in the lung and not influencing other aspects of generic systemic inflammation (although the SPD effect could be measured systemically).
Studies are beginning to address the requirements of an ideal biomarker, with therapeutic interventions for Phase II and Phase III studies in mind. For example, fibrinogen was used as a marker in a Phase II study of p38 mitogen-activated protein kinase (MAPK) inhibitors,Citation79 which effectively block the proinflammatory pathway. The treatment clearly had an effect, as determined by reducing fibrinogen levels. However, whether this pathway is merely physiological, rather than pathological, remains unknown, and hence, fibrinogen still remains uncertain as a therapeutic marker option in the long-term or short-term management of COPD. Nevertheless, this study represents a first step toward the development and assessment of new therapies using a biomarker, perhaps with a cardiovascular benefit (in those with this comorbidity), rather than a pulmonary one.
Other inflammatory markers are measurable and elevated in the plasma, and although none have proven to be specific, an alternative has been to measure several to develop a multicomponent approach.Citation80 Using the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) cohort,Citation80 the authors measured several blood biomarkers that were increased in COPD patients compared with healthy smokers and nonsmokers. The white blood cell (WBC) count, CRP, and fibrinogen levels fitted this criterion, as did IL-6, IL-8, and TNFα. The proportion outside the 95th percentile of nonsmokers was networked at baseline and at 1-year follow-up to determine consistency. A subgroup (16% of the patients) with persistently elevated inflammatory markers (two or more) was identified who had a higher incidence of exacerbations (cause or effect?) and worse survival.
This approach of selecting certain markers or traits to identify patient groups with common features is the basis for “cluster analysis”, which may be informative but clearly depends on the traits or factors selected and always needs validation in a second patient cohort; such studies are awaited.
The ECLIPSE cohort has been used to study a large number of other potential biomarkers. In 2011 Dickens et al reported on the stability of 34 blood biomarkers over 3 months.Citation81 The importance of stability is that single measurements could be informative of prognosis and particularly, may reflect the underlying disease activity that leads to progressive deterioration. Fifteen of the markers were different in COPD compared with the controls, and four reflected patients who had exacerbations between baseline and follow-up (CRP, fibrinogen, IL-6, and SPD), but few were classified as stable and hence useful as markers of baseline disease activity (fibrinogen, Clara cell protein [CC]16, and SPD). Although there were weak correlations of the other markers with spirometry, fibrinogen, which did not correlate, emerged as the most promising as it was stable and was a marker of a frequent exacerbation phenotype, exercise impairment, the severity of the BODE index, and dyspnea.
However, of these “stable” markers, only CC16 showed both a relationship to baseline spirometry and the subsequent annual decline.Citation82 At present, only this protein and the Aα-Val360 (see Elastase and other proteinases section) biomarker have shown such characteristics. CC16 is an immunosuppressant and antioxidant produced in the airways. The fact that it is elevated in COPD suggests it is reflective of the disease, rather than causative; however, it is possible that its measurement in serum (like SPD) reflects leakage from the lung due to local inflammation, which may be the generic process associated with, rather than causing, decline. Nevertheless, this series of studies demonstrates several of the steps needed to validate a biomarker, which if subsequently implicated in the pathophysiology, will also direct thinking to new specific therapeutic targets.
SPD levels in blood are also raised in COPD and respond to steroid therapy as indicated above. Since low SPD levels in the lung have been implicated in the pathophysiology of COPD,Citation83 are reduced in BAL in COPD,Citation84 and relate to spirometry,Citation85 it fulfils more of the pathophysiological role than does CC16. However, although SPD responds to oral steroids with a decrease in plasma levels, it is unknown what happens in the lung in the same patients. Nevertheless, in a separate study, COPD was associated with low lung levels of SPD, and inhaled steroid therapy was associated with higher levels,Citation84 suggesting that SPD is a good marker of disease and response to treatment. Subsequently, a polymorphism of the SPD gene was identified that also relates to COPDCitation86 and that is reflected in the level of SPD – at least in the serum (transcription and lung levels are currently unknown). This latter point is of importance: whereas this appears to “close the loop”, it should be noted that a functional polymorphism of SPD that is of pathophysiological importance should result in low lung levels of SPD, but the presence of airway inflammation and leak may, counterintuitively, lead to high blood levels. Nevertheless, the studies do suggest the potential for local therapy with SPD in COPD and that it is certainly worth pursuing.
Sampling issues
Studies of the pathophysiology of COPD are made all the more complex because of the different pathological phenotypes and the fact that the site of damage/repair is in the local tissues. Clearly this is not an issue if the biomarker is either genetic (as in AATD), or systemically reflects the disease process or specifically samples the disease area. To date, biomarkers have been studied in exhaled breath condensate, spontaneous or induced sputum, BAL, or serum/plasma. All of these have inherent problems in interpretation, especially when using high-throughput methodology.
Firstly, many commercially available assays have not been validated using the biological medium collected and processed for the measurement (especially airway secretions) and can produce inaccurate results.Citation87 Secondly, the mediator measurement (especially in exhaled breath condensate) may be detectable but often below the lower limit of quantification.Citation88 Thirdly, the biological medium may not specifically sample the area where the disease process is occurring. For instance, sputum or induced sputum is unlikely to represent samples derived from the small airways or alveoli, and BAL does not exclusively sample the small airways or alveolar region. In addition, the variability between samples from the lung is often wider than the differences between subjects or clinical states. For instance, markers in spontaneous sputum may have a day to day variability of up to 200%, for many reasons.Citation89 Obtaining samples sequentially and averaging the results drastically reduces this variability, if the patient is stable;Citation89 however, this approach cannot easily be applied to invasive procedures, such as BAL or induced sputum, since the introduction of saline or hypertonic saline itself is proinflammatory, and several days are needed between sampling to enable a return to the stable state. However, serum or plasma stability is much more reliable, at least for some biomarkers.Citation81 Pathological specimens provide an opportunity to sample individual cells and their nature/metabolic activity, by laser capture methodology, but other sampling techniques provide varying information from normal and abnormal tissue and may not reflect the pathological area unless performed on lung resection material within defined pathological areas. Finally, mediators measured in blood samples may reflect processes occurring in the interstitium, recirculated from lymph but not necessarily the airways. The subsequent volume of distribution, not only dilutes mediators but also, clears them by appropriate receptor binding.Citation90 Even then, studies like proteomics and metabolomics will differ in plasma compared with serum, due to coagulation and also, by sample collection, distribution, processing, and storage. Careful matching with appropriate controls and interpretation of data is therefore essential in all studies but is rarely done or reported, making the comparison between studies relatively tenuous.
Exacerbations
Exacerbations are an important contributor to health status, health care utilization, and death, particularly if leading to hospitalization. The definition of this symptom-led event, its complexity, and the broad approach to treatment and prevention has generated a search for an objective measure of an exacerbation and therapeutic guidance.
In general, exacerbations are events that are associated with an increase in inflammation, both in the lung and systematically.Citation91 However, in general, the events may reflect a bacterial or viral cause or be related to other causes of increased airway obstruction or may even reflect the destabilization of other organ systems, such as in associated cardiovascular decompensation.Citation92 The main decision required for such events is whether to increase bronchodilator therapy alone, to treat with corticosteroids or antibiotics, or both. Of these, the use of antibiotics has received most attention because overuse is likely to lead to the continued development of bacterial resistance.
Serum procalcitonin is normally produced by neuroendocrine cells of the thyroid and lung but is also produced elsewhere in the body, in response to bacterial infection. For this reason, it has been used to predict bacterial infections and outcome in patients with sepsis.Citation93 However in general this approach has been unsuccessful in COPD.Citation94.Citation95 Serum amyloid A has also been suggested for this as it is increased during exacerbations with a viral or bacteriological aetiology;Citation96 however, it failed to differentiate them, despite reflecting severity of the episode, and hence would not facilitate treatment guidance.
To date, the best two markers that influence therapy are symptom deterioration with known or current eosinophilia, which likely benefit from steroid therapy,Citation65 or sputum color. The latter reflects the presence or absence of a large neutrophilic load, imparting various shades of green. A change in color reflects an increase or decrease in sputum neutrophilia. The color can be characterized at least semiobjectively by both the patient and health care worker, using a standard color chart. In the initial study using this approach in primary care, there was high sensitivity to bacteria isolated from sputum and in particular, an increase relating to the colonizing microbial load, even in the stable clinical state.Citation44 This approach, using sputum color, enabled antibiotics to be withheld without detriment from patients with white or colorless sputumCitation74 and also enabled monitoring of the antibiotic response by the patients.Citation97 Subsequent studies have confirmed the robust nature of this approach in identifying patients with significant colonizationCitation98 and also, that withholding antibiotics from those without yellow or green sputum had no adverse effects.Citation94 Interestingly the authors found that CRP and not procalcitonin was increased in patients with purulent sputum, suggesting that CRP may be a better discriminator for bacterial infection. Clearly, further clinical trials are indicated to consolidate this approach.
Technology platforms and the “omics” revolution
In recent years, the ability to obtain multiple assay kits commercially and the development of the proteomics, metabolomics, transcriptomics, and genomics approaches to patient assessment has led to an overwhelming plethora of data. This does lead to some technical issues: the assay may not have been validated for the biological sample or the processing methodology used may lead to spurious and unrecognized effects on the result, both within and between patients. In addition, the data generated has a sensitivity that far outweighs our current ability to phenotype patients sufficiently from a clinical, physiological, and radiological viewpoint. It may be that this approach will lead to a pathophysiological classification of COPD that negates clinical phenotyping, although this remains to be determined and would have to be validated against the clinical “gold standard”.
Proteomics (with 75 citations in the past year) is based on the identification of COPD proteins in biological fluids by a multitude of techniques, which produce a two-dimensional spectrum of data () to determine the similarities or differences between subjects. This demonstrates the amount of data that can be generated by the “omics” approach. The benefit is that this is not a “hypothesis-led” approach, and new data can emerge as “hypothesis generating”. Nevertheless, such an approach can lead to false-positive and false-negative results, and new statistical approaches to the data are needed.Citation99
Figure 5 A two dimensional proteomics read out. Each dot/blot represents a single peptide. Changes in density/presence are considered as biomarkers of the disease progress or activity.
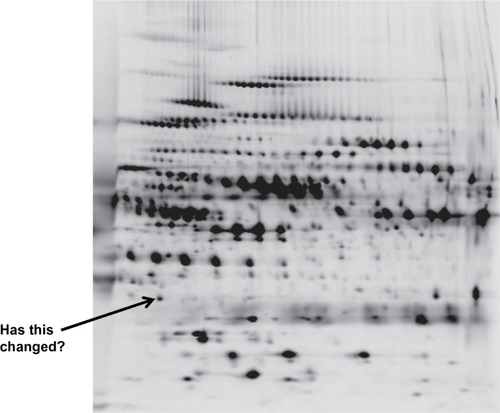
Indeed it has even been suggested that combining the data from different “omics” platforms, as part of an integrated system biological approach to understanding lung diseases, may be necessary to make progress.Citation100 The methodology is advancing at great pace. The review by Nicholas et alCitation101 defines the principles and aims of the methodology and is worth reading, for those interested.
Less has been published on metabolomics, but in a small study of smokers with emphysema, the assessment of 3,534 metabolite markers was able to separate smokers with and without emphysema,Citation102 and the top 12 were used for predictive models, some of which produced clear separation (as good as CT scan?).
Genetic studies have dominated the COPD world since the early 2000s, with both a candidate gene approach and genome-wide association studies. The data have aimed at detecting the genetic markers of COPD predisposition, thereby leading to new therapies based on gene function (over- or underactive) or a preventative approach to the disease(s). Many of the genetic markers failed to identify polymorphisms that influence the targeted gene function, but associations with COPD or its features suggested the marker was in linkage disequilibrium, probably with a nearby functional abnormality. Such susceptibility markers known to be associated with functional abnormalities include matrix metallopeptidase 9 (MMP9) and apical emphysema,Citation103 TNFα and bronchitis,Citation104 superoxide dismutase and emphysema,Citation105 iron-responsive element binding protein 2 (IREB2) and COPD,Citation106 and IL-13 promoter polymorphisms and an interaction with cigarette smoking on FEV1.Citation107 Nevertheless, such associations represent only minor contributors to the COPD complex that could reflect either the superficial phenotyping of patients or indeed the complexity of the disease(s).Citation108
Such studies do reflect potential susceptibility genes and hence, pathophysiological pathways, but how or whether they reflect the rate of progression or long-term outcomes has yet to be determined. Nevertheless, in other diseases, treatments have been influenced by genetic studies, suggesting the same may follow for COPD, at least for those associated with a functional contribution.Citation109 It may be that a panel of genes or a panel of gene transcripts may provide further insight into progression or the response to treatment, although far from personalized medicine.Citation110 Interestingly, a recent studyCitation111 has suggested that a polymorphism in the a disintegrin and metalloprotease domain 33 (ADAM33) gene is associated with all-cause and cardiovascular mortality in COPD. The group of proteins encoded by this gene domain play important roles in cell adhesion, migration, and proteolysis, and ADAM33 has also been implicated in the rapid decline in lung function and development of COPD.Citation112,Citation113 Whether this reflects a functional abnormality and hence, a treatment target needs to be determined.
Summary
Biomarker discovery has become a rapidly expanding field of research primarily to develop easy and early readouts for delivery of Phase II clinical studies and a shortening of pivotal Phase III studies that displace conventional markers like FEV1. A variety of methodologies and sample collections have been undertaken, measuring things as diverse as the mechanics of the lung, patient symptomatology, markers of exposure (such as inhaled oxidants from cigarette smoke), markers of tissue damage (connective tissue degradation products), number and function of implicated cells, generalized inflammatory markers, cell-activation products, underlying proteins (their function, metabolism, and source), and the genetic background of the patients. These have involved a variety of biological samples collected in different ways, all with both benefits and drawbacks and all requiring caution in interpretation, as summarized in . However, the identification and understanding of such markers, either in a hypothesis-testing or hypothesis-generating approach, may prove sufficiently informative for understanding the disease process and for developing new therapeutic strategies. Nevertheless, without accurate patient characterization, biomarker validation, and an understanding of the issues that likely influence the measurements (including current therapy and dissecting cause and effect), this process may prove to be both fruitless and confusing.
Table 1 Biomarkers in context
Disclosure
The author has lectured widely as part of pharmaceutical sponsored symposia, sat on numerous advisory boards for drug design and trial implementation, and received noncommercial grant funding. The author reports no other conflicts of interest in this work.
References
- SevenoaksMJStockleyRAChronic Obstructive Pulmonary Disease, inflammation and co-morbidity – a common inflammatory phenotype?Respir Res20067701916390543
- CoteCGSurrogates of mortality in chronic obstructive pulmonary diseaseAm J Med200611910 Suppl 1S54S6216843086
- DonaldsonGCWedzichaJACOPD exacerbations 1: EpidemiologyThorax200661216416816443707
- ClarkKDWardrobe-WongNElliottJJGillPTTaitNPSnashallPDPatterns of lung disease in a “normal” smoking population: are emphysema and airflow obstruction found together?Chest2001120374374711555504
- JonesPWHealth status and the spiral of declineCOPD200961596319229709
- DawkinsPADawkinsCLWoodAMNightingalePGStockleyJAStockleyRARate of progression of lung function impairment in alpha1-antitrypsin deficiencyEur Respir J20093361338134419164359
- Domingo-SalvanyALamarcaRFerrerMHealth-related quality of life and mortality in male patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2002166568068512204865
- NishimuraKIzumiTTsukinoMOgaTDyspnea is a better predictor of 5-year survival than airway obstruction in patients with COPDChest200212151434144012006425
- GerardiDALovettLBenoit-ConnorsMLReardonJZZuWallackRLVariables related to increased mortality following out-patient pulmonary rehabilitationEur Respir J1996934314358730000
- LandboCPrescottELangePVestboJAlmdalTPPrognostic value of nutritional status in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med199916061856186110588597
- CelliBRCoteCGMarinJMThe body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary diseaseN Engl J Med2004350101005101214999112
- CasanovaCde TorresJPAguirre-JaímeAThe progression of chronic obstructive pulmonary disease is heterogeneous: the experience of the BODE cohortAm J Respir Crit Care Med201118491015102121836135
- MartinezFJHanMKAndreiACNational Emphysema Treatment Trial Research GroupLongitudinal change in the BODE index predicts mortality in severe emphysemaAm J Respir Crit Care Med2008178549149918535255
- JonesPWBrusselleGDal NegroRWProperties of the COPD assessment test in a cross-sectional European studyEur Respir J2011381293521565915
- ParrDGStoelBCStolkJStockleyRAPattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairmentAm J Respir Crit Care Med2004170111172117815306534
- HarunaAMuroSNakanoYCT scan findings of emphysema predict mortality in COPDChest2010138363564020382712
- KleinJSGamsuGWebbWRGoldenJAMüllerNLHigh-resolution CT diagnosis of emphysema in symptomatic patients with normal chest radiographs and isolated low diffusing capacityRadiology199218238178211535900
- StolkJStockleyRAStoelBCRandomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptorEur Respir J201240230631222282548
- DirksenAPiitulainenEParrDGExploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiencyEur Respir J20093361345135319196813
- StolkJCooperBGStoelBRetinoid treatment of Emphysema in Patients on the Alpha-1 International Registry. The REPAIR study: study design, methodology and quality control of study assessmentsTher Adv Respir Dis20104631933220926506
- GalbánCJHanMKBoesJLComputed tomography-based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progressionNat Med201218111711171523042237
- TanabeNMuroSOgumaTComputed tomography assessment of pharmacological lung volume reduction induced by bronchodilators in COPDCOPD20129440140822509949
- Martínez-GarcíaMAde la Rosa CarrilloDSoler-CataluñaJJPrognostic value of bronchiectasis in patients with moderate-to-severe chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2013187882383123392438
- GuerraSSherrillDLVenkerCCeccatoCMHalonenMMartinezFDChronic bronchitis before age 50 years predicts incident airflow limitation and mortality riskThorax2009641089490019581277
- de OcaMMHalbertRJLopezMVThe chronic bronchitis phenotype in subjects with and without COPD: the PLATINO studyEur Respir J2012401283622282547
- CalverleyPMBoonsawatWCsekeZZhongNPetersonSOlssonHMaintenance therapy with budesonide and formoterol in chronic obstructive pulmonary diseaseEur Respir J200322691291914680078
- CalverleyPMRabeKFGoehringUMKristiansenSFabbriLMMartinezFJM2-124 and M2-125 study groupsRoflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trialsLancet2009374969168569419716960
- KuhnCYuSYChraplyvyMLinderHESeniorRMThe induction of emphysema with elastase. II. Changes in connective tissueLab Invest1976344372380177809
- LaurellCBErikssonSThe electrophoretic alpha 1-globulin pattern of serum in alpha 1-antitrypsin deficiencyScand J Clin Lab Invest196315132140
- SeniorRMTegnerHKuhnCOhlssonKStarcherBCPierceJAThe induction of pulmonary emphysema with human leukocyte elastaseAm Rev Respir Dis19771163469475900634
- KaoRCWehnerNGSkubitzKMGrayBHHoidalJRProteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamstersJ Clin Invest1988826196319733198760
- ShapiroSDEndicottSKProvinceMAPierceJACampbellEJMarked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbonJ Clin Invest1991875182818342022748
- FregoneseLFerrariFFumagalliMLuisettiMStolkJIadarolaPLong-term variability of desmosine/isodesmosine as biomarker in alpha-1-antritrypsin deficiency-related COPDCOPD20118532933321793711
- LuisettiMMaSIadarolaPDesmosine as a biomarker of elastin degradation in COPD: current status and future directionsEur Respir J20083251146115718978133
- ViglioSIadarolaPLupiAMEKC of desmosine and isodesmosine in urine of chronic destructive lung disease patientsEur Respir J20001561039104510885422
- Skjøt-ArkilHClausenRERasmussenLMAcute Myocardial Infarction and Pulmonary Diseases Result in Two Different Degradation Profiles of Elastin as Quantified by Two Novel ELISAsPLoS One201386e6093623805173
- LindbergCAEngströmGde VerdierMGTotal desmosines in plasma and urine correlate with lung functionEur Respir J201239483984521965222
- TurinoGMMaSLinYYCantorJOLuisettiMMatrix elastin: a promising biomarker for chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2011184663764121757624
- MorrisonHMWelgusHGStockleyRABurnettDCampbellEJInhibition of human leukocyte elastase bound to elastin: relative ineffectiveness and two mechanisms of inhibitory activityAm J Respir Cell Mol Biol1990232632692310584
- StockleyRABurnettDAlpha1-antitrypsin and leukocyte elastase in infected and non-infected sputumAm Rev Respir Dis197912010811086315741
- HillATCampbellEJBayleyDLHillSLStockleyRAEvidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with alpha(1)-antitrypsin deficiency (PiZ)Am J Respir Crit Care Med199916061968197510588615
- StockleyRAHillSLMorrisonHMStarkieCMElastolytic activity of sputum and its relation to purulence and to lung function in patients with bronchiectasisThorax19843964084136565423
- JacksonAHHillSLAffordSCStockleyRASputum sol-phase proteins and elastase activity in patients with cystic fibrosisEur J Respir Dis19846521141246421611
- HillATCampbellEJHillSLBayleyDLStockleyRAAssociation between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitisAm J Med2000109428829510996579
- SapeyEStockleyRANeutrophilsBarnesPJChronic Obstructive Pulmonary Disease: Cellular and Molecular Mechanisms198New York, NYTaylor and Francis Group2005133170
- SindenNJStockleyRAProteinase 3 activity in sputum from subjects with alpha-1-antitrypsin deficiency and COPDEur Respir J20134151042105022936713
- BurnettDReynoldsJJWardRVAffordSCStockleyRATissue inhibitor of metalloproteinases and collagenase inhibitory activity in lung secretions from patients with chronic obstructive bronchitis: effect of corticosteroid treatmentThorax198641107407453787506
- BurnettDCrockerJStockleyRACathepsin B-like cysteine proteinase activity in sputum and immunohistologic identification of cathepsin B in alveolar macrophagesAm Rev Respir Dis198312859159196357015
- StockleyRARennardSIRabeKCelliBProteinases and COPDSullivanALStockleyRAChronic Obstructive Pulmonary DiseaseOxfordBlackwell Publishing Ltd2008349366
- GadekJEKleinHGHollandPVCrystalRGReplacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjectsJ Clin Invest1981685115811657028785
- StockleyRABayleyDLUnsalIDowsonLJThe effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiencyAm J Respir Crit Care Med2002165111494149812045122
- CarterRIMumfordRATreonzeKMThe fibrinogen cleavage product Aα-Val360, a specific marker of neutrophil elastase activity in vivoThorax201166868669121617168
- CarterRIUngursMJMumfordRAStockleyRAAα-Val360: a marker of neutrophil elastase and COPD disease activityEur Respir J2013411313822523359
- ButtleDJAbrahamsonMBurnettDHuman sputum cathepsin B degrades proteoglycan, is inhibited by alpha 2-macroglobulin and is modulated by neutrophil elastase cleavage of cathepsin B precursor and cystatin CBiochem J1991276Pt 23253311710889
- GeraghtyPRoganMPGreeneCMAlpha-1-antitrypsin aerosolised augmentation abrogates neutrophil elastase-induced expression of cathepsin B and matrix metalloprotease 2 in vivo and in vitroThorax200863762162618250185
- OkadaYNakanishiIActivation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 (‘gelatinase’) by human neutrophil elastase and cathepsin GFEBS Lett198924923533562544455
- ChurgAZhouSWrightJLSeries “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPDEur Respir J201239119720921920892
- StănescuDSannaAVeriterCAirways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophilsThorax19965132672718779129
- SubramanianDRJenkinsLEdgarRQuraishiNStockleyRAParrDGAssessment of pulmonary neutrophilic inflammation in emphysema by quantitative positron emission tomographyAm J Respir Crit Care Med2012186111125113222837375
- DamianoVVTsangAKucichUImmunolocalization of elastase in human emphysematous lungsJ Clin Invest19867824824933525610
- BurnettDChambaAHillSLStockleyRANeutrophils from subjects with chronic obstructive lung disease show enhanced chemotaxis and extracellular proteolysisLancet198728567104310462889963
- WoolhouseISBayleyDLLalorPAdamsDHStockleyRAEndothelial interactions of neutrophils under flow in chronic obstructive pulmonary diseaseEur Respir J200525461261715802333
- SapeyEStockleyJAGreenwoodHBehavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med201118391176118621257786
- BrightlingCEMonteiroWWardRSputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trialLancet200035692401480148511081531
- BafadhelMMcKennaSTerrySBlood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trialAm J Respir Crit Care Med20121861485522447964
- PostmaDSde VriesKKoëterGHSluiterHJIndependent influence of reversibility of air-flow obstruction and nonspecific hyperreactivity on the long-term course of lung function in chronic air-flow obstructionAm Rev Respir Dis198613422762802874759
- LouhelainenNMyllärniemiMRahmanIKinnulaVLAirway biomarkers of the oxidant burden in asthma and chronic obstructive pulmonary disease: current and future perspectivesInt J Chron Obstruct Pulmon Dis20083458560319281076
- KirkhamPABarnesPJOxidative stress in COPDChest2013144126627323880677
- FischerBMPavliskoEVoynowJAPathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammationInt J Chron Obstruct Pulmon Dis2011641342121857781
- RidkerPMRifaiNRoseLBuringJECookNRComparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular eventsN Engl J Med2002347201557156512432042
- AronsonDRotermanIYiglaMInverse association between pulmonary function and C-reactive protein in apparently healthy subjectsAm J Respir Crit Care Med2006174662663216778162
- Pinto-PlataVMMüllerovaHTosoJFC-reactive protein in patients with COPD, control smokers and non-smokersThorax2006611232816143583
- DahlMVestboJZachoJLangePTybjærg-HansenANordestgaardBGC reactive protein and chronic obstructive pulmonary disease: a Mendelian randomisation approachThorax201166319720421059738
- StockleyRAO’BrienCPyeAHillSLRelationship of sputum color to nature and outpatient management of acute exacerbations of COPDChest200011761638164510858396
- PereraWRHurstJRWilkinsonTMInflammatory changes, recovery and recurrence at COPD exacerbationEur Respir J200729352753417107990
- DuvoixADickensJHaqIBlood fibrinogen as a biomarker of chronic obstructive pulmonary diseaseThorax201368767067622744884
- LomasDASilvermanEKEdwardsLDEvaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints Study InvestigatorsSerum surfactant protein D is steroid sensitive and associated with exacerbations of COPDEur Respir J20093419510219164344
- SinDDManSFMarciniukDDABC (Advair, Biomarkers in COPD) InvestigatorsThe effects of fluticasone with or without salmeterol on systemic biomarkers of inflammation in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2008177111207121418310480
- LomasDALipsonDAMillerBELosmapimod Study InvestigatorsAn oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary diseaseJ Clin Pharmacol201252341642422090363
- AgustíAEdwardsLDRennardSIEvaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) InvestigatorsPersistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotypePLoS One201275e3748322624038
- DickensJAMillerBEEdwardsLDSilvermanEKLomasDATal-SingerREvaluation of COPD Longitudinally to Identify Surrogate Endpoints (ECLIPSE) study investigatorsCOPD association and repeatability of blood biomarkers in the ECLIPSE cohortRespir Res20111214622054035
- VestboJEdwardsLDScanlonPDECLIPSE InvestigatorsChanges in forced expiratory volume in 1 second over time in COPDN Engl J Med2011365131184119221991892
- KnudsenLOchsMMackayRTruncated recombinant human SP-D attenuates emphysema and type II cell changes in SP-D deficient miceRespir Res200787017915009
- SimsMWTal-SingerRMKiersteinSChronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels: a cross-sectional studyRespir Res200891318226251
- SinDDLeungRGanWQManSPCirculating surfactant protein D as a potential lung-specific biomarker of health outcomes in COPD: a pilot studyBMC Pulm Med200771317922919
- ForemanMGKongXDeMeoDLPolymorphisms in surfactant protein-D are associated with chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol201144331632220448057
- WoolhouseISBayleyDLStockleyRAEffect of sputum processing with dithiothreitol on the detection of inflammatory mediators in chronic bronchitis and bronchiectasisThorax200257866767112149524
- BayleyDLAbusriwilHAhmadAStockleyRAValidation of assays for inflammatory mediators in exhaled breath condensateEur Respir J200831594394818216059
- SapeyEBayleyDAhmadANewboldPSnellNStockleyRAInter-relationships between inflammatory markers in patients with stable COPD with bronchitis: intra-patient and inter-patient variabilityThorax200863649349918057097
- SindenNJStockleyRASystemic inflammation and comorbidity in COPD: a result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidenceThorax2010651093093620627907
- AaronSDAngelJBLunauMGranulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2001163234935511179105
- BeghéBVerduriARocaMFabbriLMExacerbation of respiratory symptoms in COPD patients may not be exacerbations of COPDEur Respir J201341499399523543648
- BeckerKLSniderRNylenESProcalcitonin in sepsis and systemic inflammation: a harmful biomarker and a therapeutic targetBr J Pharmacol2010159225326420002097
- SolerNEsperattiMEwigSHuertaAAgustíCTorresASputum purulence-guided antibiotic use in hospitalised patients with exacerbations of COPDEur Respir J20124061344135322523352
- FalseyARBeckerKLSwinburneAJUtility of serum procalcitonin values in patients with acute exacerbations of chronic obstructive pulmonary disease: a cautionary noteInt J Chron Obstruct Pulmon Dis2012712713522399852
- BozinovskiSHutchinsonAThompsonMSerum amyloid a is a biomarker of acute exacerbations of chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2008177326927818006888
- WoolhouseISHillSLStockleyRASymptom resolution assessed using a patient directed diary card during treatment of acute exacerbations of chronic bronchitisThorax2001561294795311713358
- MiravitllesMMarínAMonsóEColour of sputum is a marker for bacterial colonisation in chronic obstructive pulmonary diseaseRespir Res2010115820470372
- WheelockÅMWheelockCETrials and tribulations of ‘omics data analysis: assessing quality of SIMCA-based multivariate models using examples from pulmonary medicineMol Biosyst20139112589259623999822
- AuffrayCAdcockIMChungKFDjukanovicRPisonCSterkPJAn integrative systems biology approach to understanding pulmonary diseasesChest201013761410141620525651
- NicholasBLO’ConnorCDDjukanovicRFrom proteomics to prescription-the search for COPD biomarkersCOPD20096429830319811390
- PaigeMBurdickMDKimSXuJLeeJKMichael ShimYPilot analysis of the plasma metabolite profiles associated with emphysematous Chronic Obstructive Pulmonary Disease phenotypeBiochem Biophys Res Commun2011413458859321925153
- ItoINagaiSHandaTMatrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysemaAm J Respir Crit Care Med2005172111378138216126934
- SapeyEWoodAMAhmadAStockleyRATumor necrosis factor-{alpha} rs361525 polymorphism is associated with increased local production and downstream inflammation in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2010182219219920299531
- SørheimICDeMeoDLWashkoGInternational COPD Genetics Network InvestigatorsPolymorphisms in the superoxide dismutase-3 gene are associated with emphysema in COPDCOPD20107426226820673035
- DeMeoDLMarianiTBhattacharyaSIntegration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility geneAm J Hum Genet200985449350219800047
- SadeghnejadAMeyersDABottaiMSterlingDABleeckerEROharJAIL13 promoter polymorphism 1112C/T modulates the adverse effect of tobacco smoking on lung functionAm J Respir Crit Care Med2007176874875217615386
- CooksonWOMoffattMFGenetics of complex airway diseaseProc Am Thorac Soc20118214915321543792
- HallIPStratified medicine: drugs meet geneticsEur Respir Rev201322127535723457165
- MarianiTJMartinezFChronic obstructive pulmonary disease genomics: yesterday, discovering population biomarkers; tomorrow, defining disease clustersAm J Respir Crit Care Med2013187990090223634854
- FigarskaSMVonkJMvan DiemenCCPostmaDSBoezenHMADAM33 gene polymorphisms and mortality. A prospective cohort studyPLoS One201387e6776823861802
- van DiemenCCPostmaDSVonkJMBruinenbergMSchoutenJPBoezenHMA disintegrin and metalloprotease 33 polymorphisms and lung function decline in the general populationAm J Respir Crit Care Med2005172332933315879414
- GosmanMMBoezenHMvan DiemenCCA disintegrin and metalloprotease 33 and chronic obstructive pulmonary disease pathophysiologyThorax200762324224717090574