
Abstract
Current antiretroviral (ARV) therapy for the treatment of human immunodeficiency virus (HIV-1)-infected patients provides long-term control of viral load (VL). Darunavir (DRV) is a nonpeptidomimetic protease inhibitor approved for use with a ritonavir booster (DRV/r). This study evaluated the effectiveness of DRV/r in combination with other ARV agents in routine clinical practice in Italy. In this descriptive observational study, data on utilization of DRV/r, under the conditions described in the marketing authorization, were collected from June 2009 to December 2012. Effectiveness (VL <50 copies/mL), tolerability, and durability in four patient groups (two DRV/r-experienced, one ARV-experienced DRV/r-naïve, and one ARV-naïve) were analyzed. Secondary objectives included immunological response, safety, and persistence/discontinuation rates. In total, 875 of 883 enrolled patients were included in the analysis: of these, 662 (75.7%) completed the follow-up until the end of 2012 and 213 (24.3%) withdrew from the study earlier. Initial DRV dose was 600 mg twice daily (67.1%) or 800 mg once daily (32.9%). Only 16 patients (1.8%) withdrew from the study due to virological failure. Virological response proportions were higher in patients virologically suppressed at study entry versus patients with baseline VL ≥50 copies/mL in each ARV-experienced group, while there was no consistent difference across study groups and baseline VL strata according to baseline CD4+ cell count. CD4+ cell count increased from study entry to last study visit in all the four groups. DRV/r was well tolerated, with few discontinuations due to study-emergent nonfatal adverse events (3.0% overall, including 2.1% drug-related) or deaths (3.0% overall, all non-drug-related); 35.3% of patients reported ≥1 adverse events. These observational data show that DRV/r was effective and well tolerated in the whole patient population described here. The DRV/r-containing regimen provided viral suppression in a high percentage of patients in all groups, with low rates of discontinuation due to virological failure.
Introduction
Recent advances in highly active antiretroviral (ARV) therapy (ART) regimens for the treatment of human immunodeficiency virus (HIV-1)-infected patients have led to considerable improvements in the long-term control of viral load (VL) and prevention of resistance. Current guidelines recommend the use of a ritonavir-boosted protease inhibitor (PI/r) (alongside other options, including integrase inhibitors) as one of the preferred third agents in addition to a nucleoside reverse transcriptase inhibitor backbone including tenofovir and emtricitabineCitation1–Citation3 or abacavir/lamivudine.Citation4,Citation5
Darunavir (DRV; TMC114) is a second-generation nonpeptidomimetic PI approved for use in combination with a ritonavir booster (DRV/r) (Prezista®). DRV/r is used in combination with other ARVs for the treatment of HIV-1 infection in adult patients and can be used in a variety of patients, ranging from those who are treatment-naïve to those who are highly experienced.Citation6–Citation9 The efficacy and tolerability of DRV/r have been evaluated in registrative prospective controlled clinical trials in treatment-naïveCitation10,Citation11 and treatment-experiencedCitation12–Citation15 patients with HIV-1 infection, with documented long-term efficacy and tolerability.Citation13,Citation16–Citation18 Observational data have shown good long-term persistence with therapy and tolerability of DRV/r and support the use of this treatment in combination with a number of ARV agents.Citation19–Citation24
The primary objective of this study was to evaluate the effectiveness of DRV/r by collecting data on utilization of this agent (combined with other ARVs) in routine clinical practice in Italy under the conditions described in the marketing authorization. The persistence of DRV/r in terms of both durability of virological response and number of patients remaining on treatment (discontinuation rate) was also evaluated because treatment failure is common in the real-world setting for a number of reasons, including lack of efficacy, loss of virological response, resistance to treatment, adverse reactions, drug adherence, and patient preference.Citation25 Furthermore, to investigate whether previous clinical trial dataCitation10–Citation18,Citation26–Citation28 translate into the setting of routine clinical practice,Citation21–Citation24 virological response with DRV/r in previously DRV-treated, ARV-experienced DRV-naïve, and ARV-naïve patients was assessed. Immuno-virological responses were analyzed according to VL at study entry. It is well known that a virological response is usually more difficult to achieve in patients with a high VL or a low CD4+ cell count at baseline. However, a meta-analysis of clinical studies conducted in ARV-experienced patients showed that VL reduction with DRV/r-based ART was independent of baseline VL and CD4+ cell count.Citation29 This lack of association with baseline VL and CD4+ has also been documented in a clinical study of DRV/r in treatment-naïve patientsCitation11 but was not seen in other studies.Citation28,Citation30 Therefore, this study also determined the virological response according to CD4+ cell count at study entry to better assess the impact of this parameter on the virological response in DRV-naïve patients. The safety profile of DRV/r was also analyzed.
Materials and methods
Study design and treatment
This was an observational study in HIV-1-infected patients treated with DRV/r, conducted in the routine clinical setting. This study was registered on ClinicalTrials.gov with the identifier NCT01375881. Effectiveness, tolerability, and durability data from four groups of patients with HIV-1 infection (two DRV/r-experienced and two DRV/r-naïve) were collected from June 2009 to December 2012. Group 1 included patients who had been on treatment with DRV/r since July 2007 or earlier, and who were part of the DRV/r Early Access Program (EAP) (subjects included in the EAP were heavily experienced, not achieving virological suppression on current regimen, at risk of clinical or immunological progression, and with limited or no treatment options); Group 2 included patients already receiving DRV/r in routine clinical practice, with treatment initiated after marketing authorization (July 2007) who had retrospective data from the start of DRV/r treatment available; Group 3 included ARV-experienced DRV-naïve patients; and Group 4 included ARV-naïve DRV-naïve patients.
Patients were treated with DRV/r in routine clinical practice according to the European Summary of Product Characteristics.Citation6 In compliance with the “Circolare del Ministero della Salute” dated 02/09/2002 and local guidelines on observational studies dated 20/03/2008, the medicinal product was prescribed according to the current clinical practice and in accordance with the terms of marketing authorization. For the DRV-naïve patients, assignment of a patient to DRV/r was not decided in advance by the study protocol but was selected on an individual basis according to clinical guidelines as part of current clinical practice.
Ethics
This study was approved by the local ethics committees of all participating centers, as follows:
– National Institute of Infectious Diseases (INMI) “L. Spallanzani” – Rome (Coordinating Center)
– “Umberto I” Policlinic – Rome
– “Luigi Sacco” University Hospital – Milan
– “San Raffaele” University Hospital – Milan
– “San Paolo” University Hospital – Milan
– “Tor Vergata” Policlinic – Rome
– Hospital “Spedali Civili” – Brescia
– “Cotugno” Hospital – Naples
– “Ospedali Riuniti” – Foggia
– Azienda USL n° 8 – Presidio Ospedaliero SS. Trinità – Cagliari
– Policlinico Universitario di Cagliari
– University Hospital “Amedeo di Savoia” – Turin
– Università Cattolica del Sacro Cuore – Policlinico Gemelli – Rome
– Ospedale Maggiore – Bologna
– Hospital “San Giovanni” – Rome
– University Hospital of Modena and Reggio Emilia
– “P. Giaccone” Policlinic – Palermo
– IRCCS Policlinico S. Matteo – Pavia
– Hospital “San Gerardo” – Monza
– Ospedale di Circolo – Busto Arsizio
– Hospital “Galliera” – Genova
– University Hospital “Careggi” – Florence
– A.U.S.L. Pescara – Pescara
– Manzoni Hospital – Lecco
– University Hospital – Ferrara
– Pugliese Ciaccio Hospital – Catanzaro
– Ethics Committee for Clinical Trials of Medicines of the Province of Venice – Venezia
– “S. Orsola” Policlinic – Bologna
– USSL 18 Rovigo – Rovigo
– Varese Hospital – Varese
Participants
Adult patients (≥18 years) with HIV-1 infection who initiated DRV/r treatment according to the label (ARV-experienced or ARV-naïve) or started DRV/r as part of an Italian EAP or who were already receiving DRV/r (provided that data at DRV/r start were available retrospectively) according to the European Summary of Product Characteristics of DRV were eligible for inclusion in this noninterventional study. All patients provided a signed and dated informed consent form for collection of prospective and retrospective data.
Patients with any of the following criteria were excluded from the study: known hypersensitivity to DRV/r or to any of its excipients; severe hepatic impairment (Child–Pugh class C); coadministration of agents known to interact with DRV/r; pregnancy or lactation; unable to read, understand, and sign the informed consent form; previously treated with DRV/r and discontinued for any reason; and participation in other interventional clinical studies.
Study end points and assessments
The primary objective was to evaluate the effectiveness of DRV/r used under the conditions described in the marketing authorization, in combination with other ARVs, in routine clinical practice in Italy. Effectiveness was primarily measured as virological response (defined as VL <50 copies/mL) in a snapshot (last observation carried forward [LOCF]) analysis. Secondary objectives were to evaluate the impact of treatment with DRV/r on immunological response, changes in laboratory parameters, the incidence of treatment-emergent adverse events (AEs), and the rates of persistence in the study or discontinuation for any reason or for specific reasons (notably virological failure).
The VL was measured in each individual center using the more widespread tests commercially available (eg, Abbott RealTime HIV-1 assay) as per manufacturer’s instructions.
The CD4+ cell count was performed in each center as per their clinical practice, using flow cytometry automated systems as per manufacturer’s instructions.
Virological outcomes were assessed based on a threshold value for plasma HIV-RNA VL of <50 copies/mL (as per protocol, this or a lesser value being the detection limit in all participating centers) and are reported by group, by VL at study entry (<50 or ≥50 copies/mL) within groups, and by baseline CD4+ (<200 or ≥200 cells/µL) within baseline VL and group. Patients were followed up at ~1, 3, and 6 months and then every 3 months thereafter, in accordance with routine practice.
Blood chemistry variables (liver function tests, glucose, triglycerides, and total cholesterol) were determined at study entry and at intervals of ~6 months.
Statistical analyses
Due to the explorative character of this noninterventional study, no specific statistical hypothesis was formulated to calculate sample size when the study was planned. However, a sample of approximately 900 patients was considered adequate to describe the efficacy and safety profile of DRV/r in Italian patients with HIV-1 infection.
Statistical analysis was carried out using the SAS® system, PC release 9.2 (SAS Institute Inc., Cary, NC, USA).
The baseline of the study was the start of the prospective observation, and follow-up time was, therefore, calculated from this date to the last on-study visit, that is, the end of 2012 or earlier discontinuation. All subjects with HIV-1 infection who received ≥1 dose of DRV/r after enrollment were included in the full analysis of demographic and baseline characteristics as well as efficacy and safety data (full analysis population); however, the effective sample size for specific analyses was reduced in some instances because of missing data.
Data are reported for each study group and overall. Continuous data are described using standard descriptive methods, including median and other quartiles (Q1 and Q3), mean and standard deviation (SD), and interval estimates, that is, 95% confidence interval (CI) of the mean. Categorical data are summarized using proportions of totals based on nonmissing values unless otherwise stated; exact 95% CI values of response proportions were calculated.
CD4+ levels were analyzed on a LOCF basis; summary statistics were calculated at enrollment and at last study visit, including mean difference and the associated 95% CI. To evaluate their impact on changes from baseline to last value, an analysis was performed with baseline CD4+ levels and time to last observation as covariates within each group.
Blood chemistry variables (transaminases, glucose, triglycerides, and total cholesterol) were summarized at study entry and at intervals of 24±12 weeks using data from subjects with complete observations from baseline to 72±12 weeks.
LOCF analyses of VL were performed by calculating the proportion of patients with virological response (viral suppression), that is, VL <50 copies/mL irrespective of confirmation at the last study visit (snapshot analysis), following two alternative ways to classify study discontinuations:
– Modified intention-to-treat (mITT) analysis: all study discontinuations related to medical events (virological failure, AEs, or death) or due to the patient (consent withdrawal, nonadherence or poor compliance, loss to follow-up) were considered as failures overriding the final VL value, while withdrawals for investigator’s decision unrelated to outcome (such as use of nonstandard DRV dose, enrollment in a clinical trial, completion of 12-month treatment of acute infection, or therapy simplification) and DRV/r dose modifications were not imputed as failures.
– On-treatment (OT) analysis: only virological failures were considered, study discontinuations for any other reason were not imputed as failures; follow-up was censored at the date of discontinuation.
The analyses of response based on VL <50 copies/mL were stratified by baseline VL, <50 or ≥50 copies/mL, and further (for the LOCF analysis) by baseline CD4+ level, <200 or ≥200 cells/µL. Differences in response proportions associated with these characteristics were tested within groups using Fisher’s exact test and the Cochran–Mantel–Haenszel chi-square test.
Analyses of virological response based on VL <50 copies/mL were also performed at 48 and 96 weeks. Patients who withdrew before the time point (48 or 96 weeks) for any medical or patient-related reason, as described earlier, were considered as failures in the mITT analysis, while patients withdrawn for virological failure with the last VL of ≥50 copies/mL before the time point were considered as failures in the OT analysis. In either the mITT or the OT analysis, responders were patients still on treatment at the time point with a VL <50 copies/mL, while failures were defined as a VL ≥50 copies/mL at the time point, using the VL value closest to the time point if available within ±90 days, or otherwise imputing its value as <50 or ≥50 copies/mL if both the last value before this interval and the first value after this interval were consistently so. Patients enrolled after February 2011 were excluded from the analysis at 96 weeks as they could not have reached this time point by the end of the study.
To assess the impact of low (50–399 copies/mL) VL final values on the proportions of primary response defined earlier, a secondary analysis was performed with virological response defined as VL <400 copies/mL instead of <50 copies/mL.
AEs were summarized descriptively, in particular deaths and AEs that caused study interruption. Proportions and incidence rates were calculated. The probability of a patient withdrawing from the study for specific reasons was calculated over time using Kaplan–Meier curves. Separate curves were produced for each study group representing time since entry in the prospective study.
Results
Patients
A total of 883 patients were enrolled in 36 Italian centers between June 5, 2009 and November 30, 2011. Of these, 875 were included in the full analysis set for both effectiveness and safety. Follow-up continued up to the end of 2012 or earlier discontinuation. Flow of patients through the study and number of patients in the analyzed populations and groups are shown in . Overall median study duration was 95 weeks (Q1–Q3, 74–121; mean ± SD, 93±39 weeks). Mean study duration was 115±39, 83±27, 91±50, and 87±40 weeks in Groups 1, 2, 3, and 4, respectively.
Figure 1 Flow of patients through the study.
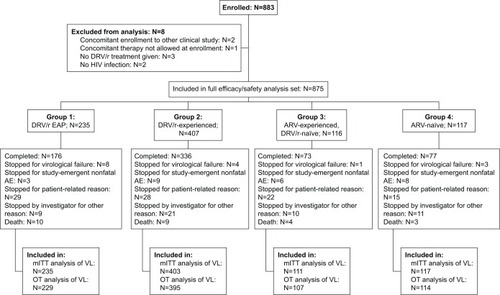
Demographic and other characteristics of the study patients at baseline (ie, start of prospective observation) are shown in . The four groups differed largely in all important anamnestic (time since diagnosis, number of previous drugs), clinical (Centers for Disease Control and Prevention stage), and laboratory (HIV-RNA VL, CD4+ cell count) characteristics as they were defined as patient groups in different phases of their ARV therapy or DRV course.
Table 1 Baseline patient characteristics and demographics (full analysis set)
The results are reported mainly descriptively by point and interval estimates within groups in the “Virological response” section; stratified analyses assessing the impact of baseline VL and CD4+ levels within groups have also been reported. Within each of Groups 1, 2, and 3, virological response was higher when baseline VL was <50 copies/mL at baseline. Overall response ratios in Groups 3 and 4 (DRV-naïve) were lower than in Groups 1 and 2 (DRV-experienced) reflecting the fact that most Group 3 patients and all Group 4 patients had high baseline VL, while most DRV-experienced patients had a baseline VL <50 copies/mL. Groups 1 and 2 were similar in the proportion of patients with baseline VL <50 copies/mL and in the response ratios within baseline VL stratum. Response ratios in Group 4 patients with baseline VL ≥50 copies/mL were higher than in Group 3 patients with baseline VL ≥50 copies/mL.
A high percentage of patients in Groups 1–3 had a long duration of HIV infection. The mean interval from DRV start to entry in the study was 41 months for patients in Group 1 and 16 months for patients in Group 2 (). At DRV start, all Group 1 patients (ex-EAP) were ARV-experienced, while of Group 2 patients, 294 were ARV-experienced, 50 ARV-naïve, and 63 unknown.
The median number of ARV active principles (other than DRV/r) reported before study start was thirteen in Group 1, eight in Group 2, and seven in Group 3. Overall, 61% of the patients received two other active ARV drugs ongoing at study start; tenofovir + emtricitabine were given together to 55.5% of the patients (91.5% of ARV-naïve), including 11.9% with additional raltegravir (). Patients initially received DRV/r 600/100 mg twice daily (bid) or 800/100 mg once daily (qd) according to their clinical conditions and current approved dose (). The majority of patients (96.1%) remained on their initial dosage throughout the study (): 557 (94.9% of 587) on 600/100 mg bid and 284 (98.6% of 288) on 800/100 mg qd. Of the 34 patients (3.9%) who changed their DRV/r dose after study entry, 30 (5.1% of 587) changed from 600/100 mg bid to 800/100 mg qd during the study and four (1.4% of 288) changed from 800/100 mg qd to 600/100 mg bid.
Table 2 Concomitant ARV treatments and DRV/r dose during the study
Virological response
Virological response proportions at the last study visit in the mITT analysis (ie, with medical and patient-related discontinuations imputed as failures) and in the OT analysis (ie, based on last VL only with no imputation), with virological failure set to VL ≥50 copies/mL as prespecified, are shown in . When assessed according to baseline VL in ARV-experienced patients (Groups 1, 2, and 3), virological response was higher in patients with baseline VL <50 copies/mL versus those with baseline VL ≥50 copies/mL (P<0.01 within each group; ). Within the same baseline VL stratum of <50 or ≥50 copies/mL, there was no consistent difference across study groups between patients with baseline CD4+ ≥200 cells/µL and CD4+ <200 cells/µL, in either the OT or the mITT analyses (P≥0.10 within each group controlling for baseline VL class and within each CD4+ cell count; ).
Table 3 Virological response (VL <50 copies/mL) at last study visit (LOCF) for the mITT and OT analyses in the four HIV-infected patient groups treated with DRV/r, according to VL and CD4+ cell count at study entry
Virological response proportions at 48 weeks and 96 weeks by mITT and OT, with virological failure set to VL ≥50 copies/mL, are reported in . As in the LOCF analysis, virological response within each ARV-experienced group was higher in subjects with baseline VL <50 copies/mL than in those with baseline VL ≥50 copies/mL, although not all differences achieved statistical significance (P<0.0001 for all Group 1 analyses; P<0.0001 for the analyses in Group 2, except P=0.15 by OT at 96 weeks; P=0.07 by mITT at 48 weeks, P=0.15 by OT at 48 weeks, P=0.04 by mITT at 96 weeks, and P=0.46 by OT at 96 weeks in Group 3). Response by mITT decreased from 48 to 96 weeks in all groups, while OT response proportions at 96 weeks compared to 48 weeks decreased in the ex-EAP patients, increased in the ARV-naïve patients, and was approximately constant in the other two groups.
Table 4 Virological response (VL <50 copies/mL) at 48 weeks and 96 weeks for the mITT and OT analyses in the four HIV-infected patient groups treated with DRV/r, according to VL at study entry
In secondary analyses with virological failure defined as VL ≥400 copies/mL instead of ≥50 copies/mL, overall response proportions by mITT were 181/235 (77.0%) in Group 1, 340/403 (84.4%) in Group 2, 72/111 (64.9%) in Group 3, and 86/115 (74.8%) in Group 4, and 218/229 (95.2%), 372/395 (94.2%), 91/107 (85.0%), and 106/112 (94.6%), respectively, by OT. Comparison with these proportions shows that in the primary OT analysis, most virological failures in Groups 1, 2, and 4 and half in Group 3 were due to final VL values between 50 and 399 copies/mL.
Only 16 withdrawals due to virological failures occurred:
– eight in patients from the EAP study (Group 1): all except one harbored multiple mutations, including DRV resistance associated mutations (RAMs), at study entry;
– four in other DRV-experienced patients (Group 2): three had only a resistance test at study entry, one of which had both reverse transcriptase (RT) and DRV RAMs; only one patient had a test both at study entry (only RT mutations) and at study end (no DRV RAMs);
– one in an ARV-experienced DRV-naïve patient (Group 3): resistance test was available only at baseline showing multiple PI mutations;
– three in ARV-naïve patients (Group 4): one patient had both study entry and follow-up resistance test with no mutations at both time points; one patient had only final resistance test with only RT mutation and no PI mutations; one patient had only study start resistance test with no mutations.
Follow-up and treatment persistence
Crude ratios of persistence in the study at the last available observation were 74.9% in Group 1, 82.6% in Group 2, 62.9% in Group 3, and 65.8% in Group 4. Reasons for study discontinuation in the four groups are shown in . In total, 213 patients (24.3%) discontinued treatment, but virological failure was the reason in only 16 patients (1.8%); 7.3% were lost to follow-up, 2.9% discontinued due to lack of compliance (including one patient who withdrew consent), 3.0% dropped out due to the onset of study-related nonfatal AEs, and 0.6% discontinued for other patient-related reasons. Other study discontinuations (5.8%) occurred because of investigators’ decisions based on conditions present at study entry or for other reasons unrelated to outcome (eg, use of a nonstandard DRV dose, enrollment in a clinical trial, completion of 12-months’ treatment for acute infection, or therapy simplification). Overall, 26 patients (3.0%) died during the study; none of the deaths were considered to be related to DRV/r.
Table 5 Reasons for discontinuation from the study
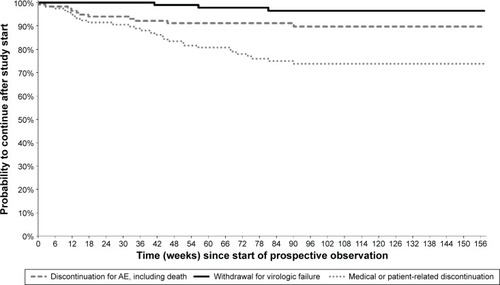
Kaplan–Meier curves showing, for each study group, the cumulative probabilities of continuing in the study or discontinuing for various reasons are presented in –. Persistence rates (100% minus the probability of discontinuing for any reason) from the start of prospective observation up to 48 weeks (or exactly 1 year) were 90.2% (89.0%) in Group 1, 90.2% (89.4%) in Group 2, 76.8% (76.0%) in Group 3, 77.9% (76.2%) in Group 4, and 86.8% (85.8%) overall. Corresponding values from the start of prospective observation up to 96 weeks (or exactly 2 years) were 79.8% (79.3%), 81.1% (80.6%), 67.4% (66.1%), 65.9% (64.7%), and 76.7% (76.0%). The Kaplan–Meier probability of withdrawal due to virological failure at 96 weeks (and identically at 2 years) from study start was 2.3% in Group 1, 1.1% in Group 2, 1.4% in Group 3, 3.5% in Group 4, and 1.8% overall, similar to the crude proportion of discontinuations for this reason at the last available observation (–).
Figure 2 Kaplan–Meier curves from start of prospective observation showing study discontinuation by reason of interruption in Group 1 (n=235) – patients who were part of the DRV/r Early Access Program (EAP).
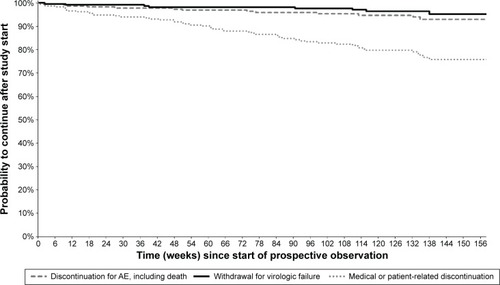
Figure 3 Kaplan–Meier curves from start of prospective observation showing study discontinuation by reason of interruption in Group 2 (n=407) – patients already receiving DRV/r in routine clinical practice.
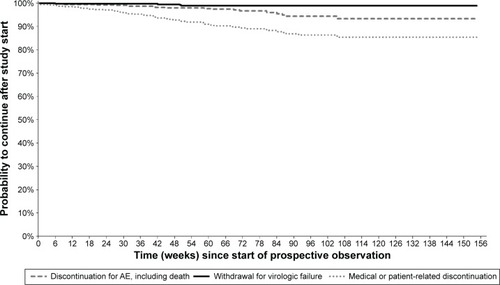
Figure 4 Kaplan–Meier curves from start of prospective observation showing study discontinuation by reason of interruption in Group 3 (n=116) – ARV-experienced DRV-naïve patients.
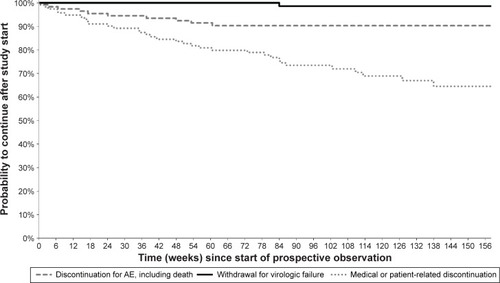
Figure 5 Kaplan–Meier curves from start of prospective observation showing study discontinuation by reason of interruption in Group 4 (n=117) – ARV-naïve patients.
Immunological recovery
The mean baseline CD4+ cell counts varied between the four groups, being considerably higher in DRV-experienced patients compared with DRV-naïve patients, especially if ARV-naïve (). CD4+ cell counts increased from baseline to last study visit in all four groups (): mean (95% CI) increases were 54 cells/µL (32, 76) in ex-EAP patients, 59 cells/µL (44, 74) in other DRV-experienced patients, 138 cells/µL (100, 176) in ARV-experienced DRV-naïve patients, and 266 cells/µL (232, 300) in ARV-naïve patients. The analysis of covariance showed that in all groups, the increase from baseline to last value was significantly greater for longer durations of observation (P<0.0001), although the relationship was not necessarily linear, and in patients with lower baseline values (P<0.05).
Table 6 CD4+ cell count at study entry and at last study visit in the four groups of HIV-infected patients treated with DRV/r (LOCF analysis)
Tolerability
Safety data from 875 patients were analyzed. DRV/r-based treatment was well tolerated, with 35.3% of patients reporting ≥1 AE. Treatment discontinuations due to AEs were few (n=26, 3.0% overall): for 18 of these, a relationship with DRV/r treatment was not excluded by the investigator (). The most frequent AEs that led to treatment discontinuation were diarrhea (n=5) and rash (n=4) ().
Median and other quartiles of serum biochemistry values for each group are reported in , only for patients with a complete set of observations at each time point: baseline (within 3 months before enrollment), 24±12, 48±12, and 72±12 weeks. Levels of the liver enzymes alanine aminotransferase and aspartate aminotransferase remained stable from study entry through 72 weeks in DRV-experienced patients, decreased slightly in ARV-experienced DRV-naïve patients, and decreased more markedly in ARV-naïve patients. Blood glucose concentrations remained stable in all groups from study entry through 72 weeks. Serum triglyceride levels decreased slightly in ex-EAP patients, remained relatively stable in Groups 2 and 3, and increased in ARV-naïve patients up to 48 weeks. Total cholesterol levels were essentially unchanged from study entry through 72 weeks in both DRV-experienced groups, while in ARV-experienced DRV-naïve patients and especially in ARV-naïve patients, median values increased at 24 weeks after which they did not increase further.
Table 7 Median (Q1–Q3) of serum biochemistry values during the study in patients with complete observations
Discussion
Although randomized, controlled clinical studies provide the highest level of evidence in terms of efficacy and tolerability for a specific treatment, observational studies are important to help clinical trial data be translated into the real-world setting because they provide information about persistence and durability in routine clinical practice that are not possible to obtain from clinical studies. Moreover, observational studies enroll an unselected population and allow long-term observation of effectiveness and safety of drug combinations. This analysis of real-world data of DRV/r treatment is the first one to include the range of patients likely to be seen in clinical practice.
The results showed that most patients, regardless of previous clinical and treatment history, were treated with a stable dose of DRV/r of 600/100 mg bid or 800/100 mg qd and remained on study for a mean of 93±39 weeks. High persistence rates were seen in all groups, especially in both DRV-experienced groups, with an overall persistence rate of >75% after 24 months.
In ARV-naïve subjects, over two-thirds (68.7%) had a baseline VL >100,000 copies/mL and 12.2% had a VL >1,000,000 copies/mL. It is, therefore, very encouraging to report that the proportion of virological suppression in these previously untreated patients was 68.4% in the mITT analysis and 79.8% in the OT analysis. Interestingly, in ARV-naïve patients, 59.8% of subjects had a CD4+ cell count <200 cells/µL and 47.9% had a count <100 cells/µL.
The results observed in the ARV-naïve DRV-naïve patients were similar to those achieved in the Phase III open-label AntiRetroviral Therapy with TMC114 Examined In Naïve Subjects (ARTEMIS) trial.Citation18 Similarly, in the 48-week once-daily Darunavir in treatment-experienced patients (ODIN) trial comparing qd versus bid DRV/r in 590 ARV-experienced HIV-infected patients, the proportions of patients achieving VL <50 copies/mL were 72.1% and 70.9%, respectively (P<0.001, qd DRV/r noninferior to bid DRV/r).Citation14
With regard to safety, this study showed that DRV/r-based treatment was well tolerated. Only 26 patients (3%) discontinued treatment due to AEs, 18 of which were deemed possibly drug-related. This indicates that DRV/r is well tolerated in the routine clinical setting and has an overall tolerability profile similar to that observed in controlled clinical trials.Citation14,Citation18
These findings are consistent with those from controlled clinical studies, showing the long-term efficacy and tolerability of DRV/r. The current results in ARV-experienced DRV-naïve patients receiving DRV/r therapy are similar to those reported in the 48-week randomized controlled Phase III TITAN study of DRV/r 600/100 mg bid.Citation15
The effectiveness, tolerability, and persistence data reported in this study are in agreement with those from other real-world studies.Citation21–Citation24 The body of evidence suggests that DRV-based therapy administered in routine care settings is associated with proportions of virological suppression similar to those seen in randomized controlled trials in most treatment-experienced patients with HIV-infectionCitation14,Citation18 This is true even in patients who have been treated for many years using several different ART regimens and who were failing current therapy due to lack of response or tolerability issues.
Limitations
The limitations of this study include the lack of a control arm and the different characteristics and origins of the study groups: two DRV-experienced groups (one from an EAP and one from routine clinical practice) and two DRV-naïve groups. However, these four groups reflect patients typically seen in daily clinical practice and may, therefore, help guide use of DRV/r in patients with HIV infection regardless of their treatment history, baseline VL, and CD4+ cell count. Another limitation to take into account is that Groups 1 and 2 (DRV-experienced subjects) excluded patients who eventually discontinued DRV/r due to virological failure or drug intolerance prior to study start (this was an exclusion criterion because of the difficulties associated with collecting clinical data retrospectively).
Conclusion
These data from a routine clinical setting showed that DRV/r was effective and well tolerated in all HIV-infected patient groups, when used in either initial or switch strategies. In all groups, DRV/r-based ART provided effective viral suppression with long-lasting durability and a low rate of withdrawals due to virological failure. The overall proportion of failures, also including discontinuation for any reason, and the tolerability profile of DRV/r were favorable and similar to that reported in controlled clinical trials.Citation11–Citation15
Acknowledgments
The authors would like to thank all the study investigators (A Antinori, A d’Arminio Monforte, P Meraviglia, G Rizzardini, A Lazzarin, P Nasta, C Mussini, T Quirino, R Cauda, G Sterrantino, A Chirianni, A Gori, M Borderi, G Di Perri, N Abrescia, I Mezzaroma, P Grossi, P Caramello, PE Manconi, F Di Sora, B Grisorio, G Parruti, P Bonfanti, G Angioni, L Minoli. S Rusconi, E Colella, G Penco, M Andreoni, L Titone, A Cattelan, L Cosco, L Sighinolfi, E Raise, and G Verucchi) and all the patients who consented to be part of the study. Medical writing assistance was provided by Mary Hines of Springer Healthcare Communications. This assistance was funded by Janssen-Cilag SpA (Italy).
Disclosure
AA has received honoraria for consultancy with Gilead Sciences, ViiV Healthcare, Merck Sharp & Dohme, Janssen-Cilag, Abbvie, and Bristol-Myers Squibb and has also received research grants from Gilead Sciences, Bristol-Myers Squibb, Janssen-Cilag, and ViiV Healthcare. AdAM has been involved in advisory boards supported by Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, and Merck Sharp & Dohme. SR has received research funding from Pfizer and Janssen-Cilag and has been involved in advisory boards or educational courses supported by Abbvie, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, now ViiV Healthcare, Merck Sharp & Dohme, and Janssen-Cilag. NG has been advisor for Gilead Sciences, AbbVie, and Janssen-Cilag, received speakers’ honoraria from Gilead Sciences, ViiV Healthcare, Bristol-Myers Squibb, Merck Sharp & Dohme, Roche, AbbVie, Boehringer Ingelheim, and Janssen-Cilag, and support for travel to meetings from Gilead Sciences, Bristol-Myers Squibb, AbbVie, Janssen-Cilag, Merck Sharp & Dohme, Roche, and ViiV Healthcare. CM has been involved in advisory boards or educational courses supported by Abbvie, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, now ViiV Healthcare, Merck Sharp & Dohme, and Janssen-Cilag. TB has received research funding and/or honoraria from Merck Sharp & Dohme, Bristol-Myers Squibb, and Janssen-Cilag. GA has received honoraria for the statistical analysis of study data from Janssen-Cilag. DM and RT are employees of Janssen-Cilag SpA, Italy. PM, AC, and EC report no conflicts of interest in this work.
References
- US Department of Health and Human Services [webpage on the Internet]Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents: US Department of Health and Human Services (HHS) Panel on Antiretroviral Guidelines for Adults and Adolescents (a Working Group of the Office of AIDS Research Advisory Council)2014 [updated May 1; cited October 7, 2014]. Available from: http://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdfAccessed March 1, 2016
- GazzardBGAndersonJBabikerABHIVA Treatment Guidelines Writing GroupBritish HIV Association guidelines for the treatment of HIV-1-infected adults with antiretroviral therapy 2008HIV Med2008956360818826546
- WilliamsIChurchillDAndersonJBritish HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2012 (Updated November 2013. All changed text is cast in yellow highlight.)HIV Med201415suppl 1185
- Ministero della Salute [webpage on the Internet]Linee Guida Italiane sull’utilizzo dei farmaci antiretrovirali e sulla gestione diagnostico-clinica delle persone con infezione da HIV 2013 [updated November; cited November 25, 2014] Available from: http://www.salute.gov.it/imgs/C_17_pubblicazioni_2074_allegato.pdfAccessed March 1, 2016
- European AIDS Clinical Society [webpage on the Internet]EACS Guidelines Version 7.0 2014 [updated October; cited 2014 November 25]. Available from: http://www.eacsociety.org/Portals/0/Guidelines_Online_131014.pdfAccessed March 1, 2016
- Janssen-Cilag International NVPrezista (Darunavir). Summary of Product Characteristics 2016 [updated December 12, 2015] Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000707/WC500041756.pdfAccessed March 18, 2016
- PhungBCYeniPDarunavir: an effective protease inhibitor for HIV-infected patientsExpert Rev Anti Infect Ther2011963164321692667
- McKeageKPerryCMKeamSJDarunavir: a review of its use in the management of HIV infection in adultsDrugs20096947750319323590
- DeeksEDDarunavir: a review of its use in the management of HIV-1 infectionDrugs2014749912524338166
- OrtizRDejesusEKhanlouHEfficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48AIDS2008221389139718614861
- MillsAMNelsonMJayaweeraDOnce-daily darunavir/ritonavir vs. lopinavir/ritonavir in treatment-naive, HIV-1-infected patients: 96-week analysisAIDS2009231679168819487905
- PozniakAOpravilMBeattyGHillAde BethuneMPLefebvreEEffect of baseline viral susceptibility on response to darunavir/ritonavir versus control protease inhibitors in treatment-experienced HIV type 1-infected patients: POWER 1 and 2AIDS Res Hum Retroviruses2008241275128018844462
- ArastehKYeniPPozniakAEfficacy and safety of darunavir/ritonavir in treatment-experienced HIV type-1 patients in the POWER 1, 2 and 3 trials at week 96Antivir Ther20091485986419812449
- CahnPFourieJGrinsztejnBWeek 48 analysis of once-daily vs. twice-daily darunavir/ritonavir in treatment-experienced HIV-1-infected patientsAIDS20112592993921346512
- MadrugaJVBergerDMcMurchieMTITAN Study GroupEfficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV-infected patients in TITAN: a randomised controlled phase III trialLancet2007370495817617272
- OrkinCDeJesusEKhanlouHFinal 192-week efficacy and safety of once-daily darunavir/ritonavir compared with lopinavir/ritonavir in HIV-1-infected treatment-naive patients in the ARTEMIS trialHIV Med201314495923088336
- BanhegyiDKatlamaCda CunhaCAWeek 96 efficacy, virology and safety of darunavir/r versus lopinavir/r in treatment-experienced patients in TITANCurr HIV Res20121017118122339125
- LathouwersEDe MeyerSDierynckIVirological characterization of patients failing darunavir/ritonavir or lopinavir/ritonavir treatment in the ARTEMIS study: 96-week analysisAntivir Ther2011169910821311113
- PodzamczerDImazAPerezIKIDAR Study GroupAbacavir/lamivudine plus darunavir/ritonavir in routine clinical practice: a multicentre experience in antiretroviral therapy-naive and -experienced patientsJ Antimicrob Chemother2014692536254024833755
- MenzaghiBRicciECarenziLSafety and durability in a cohort of HIV-1 positive patients treated with once and twice daily darunavir-based therapy (SCOLTA Project)Biomed Pharmacother20136729329823433852
- BeneaOEStreinu-CercelADorobatCEfficacy and safety of darunavir (Prezista(R)) with low-dose ritonavir and other antiretroviral medications in subtype F HIV-1 infected, treatment-experienced subjects in Romania: a post-authorization, open-label, one-cohort, non-interventional, prospective studyGerms20144596925276665
- GatheJExperience with darunavir in HIV-infected adults enrolled in a US expanded access program: results from a single centerCurr Med Res Opin20082476977318234149
- RibeiroKMBiscioneFMWestinMRMachadoDPGrecoDBTupinambasUVirologic and immunologic effectiveness of darunavir-based salvage therapy in HIV-1-infected adults in a Brazilian clinical practice setting: results of a multicenter and retrospective cohort studyBraz J Infect Dis2014181723916454
- WilligJHAbanINevinCRDarunavir outcomes study: comparative effectiveness of virologic suppression, regimen durability, and discontinuation reasons for three-class experienced patients at 48 weeksAIDS Res Hum Retroviruses2010261279128520961276
- KimHPerelsonASViral and latent reservoir persistence in HIV-1-infected patients on therapyPLoS Comput Biol20062e13517040122
- MolinaJMCohenCKatlamaCTMC114-C208 Study Group; TMC114-C215 Study GroupSafety and efficacy of darunavir (TMC114) with low-dose ritonavir in treatment-experienced patients: 24-week results of POWER 3J Acquir Immune Defic Syndr200746243117621237
- De MeyerSMSpinosa-GuzmanSVangeneugdenTJde BethuneMPMirallesGDEfficacy of once-daily darunavir/ritonavir 800/100 mg in HIV-infected, treatment-experienced patients with no baseline resistance-associated mutations to darunavirJ Acquir Immune Defic Syndr20084917918218769351
- ClotetBBellosNMolinaJMPOWER 1 and 2 Study GroupsEfficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trialsLancet20073691169117817416261
- BerhanABerhanYVirologic response to tipranavir-ritonavir or darunavir-ritonavir based regimens in antiretroviral therapy experienced HIV-1 patients: a meta-analysis and meta-regression of randomized controlled clinical trialsPLoS One20138e6081423593314
- RaffiFBabikerAGRichertLNEAT001/ANRS143 Study GroupRitonavir-boosted darunavir combined with raltegravir or tenofovir-emtricitabine in antiretroviral-naive adults infected with HIV-1: 96 week results from the NEAT001/ANRS143 randomised non-inferiority trialLancet20143841942195125103176