
Abstract
Purpose
Gastroesophageal reflux disease involves the reflux of gastric and/or duodenal content into the esophagus. Prokinetic therapies, such as the selective 5-hydroxytryptamine receptor 4 agonist revexepride, may aid gastric emptying. This Phase I study evaluated the pharmacokinetics and excretion pathways of [14C]revexepride in healthy individuals using a microtracer approach with accelerator mass spectrometry.
Participants and methods
Six healthy men received a single oral dose of 2 mg [14C]revexepride containing ~200 nCi of radioactivity; blood, urine, and fecal samples were collected over a 10-day period.
Results
Almost 100% of 14C was recovered: 38.2%±10.3% (mean ± standard deviation) was recovered in urine, and 57.3%±0.4% was recovered in feces. Blood cell uptake was low, based on the blood plasma total radioactivity ratio of 0.8. The mean revexepride renal clearance was 8.6 L/h, which was slightly higher than the typical glomerular filtration rate in healthy individuals. Time to reach maximal concentration was 1.75±1.17 hours (mean ± standard deviation). No safety signals were identified.
Conclusion
This study demonstrated that revexepride had rapid and moderate-to-good oral absorption. Excretion of radioactivity was completed with significant amounts in feces and urine. Renal clearance slightly exceeded the typical glomerular filtration rate, suggesting the involvement of active transportation in the renal tubules.
Introduction
Gastroesophageal reflux disease (GERD) is a common condition, with an estimated prevalence of 9%–26% in Europe and 18%–28% in North America.Citation1 The condition develops when reflux of stomach contents causes troublesome symptoms and/or complications.Citation2 The classic symptoms of GERD are heartburn and regurgitation,Citation2 which may be accompanied by chest pain and dysphagia. GERD significantly affects health-related quality of life and may lead to significant complications, such as ulceration, Barrett’s esophagus, and respiratory problems (eg, pneumonia), if left untreated.Citation2
Standard therapy for patients with GERD is with histamine-2-receptor antagonists or proton pump inhibitors (PPIs).Citation3 PPIs are effective in the majority of patients, but up to 40% of individuals do not have their symptoms fully controlled, despite PPIs adequately controlling acid exposure in most of these patients.Citation4,Citation5 A different mechanism, such as dysmotility, may be responsible for these remaining symptoms.Citation6,Citation7 Dysmotility can result in impaired lower esophageal sphincter function, reduced esophageal motility, and slowing of gastric emptying.Citation8 It is thought that the reduced responsiveness of the lower esophageal sphincter to acetylcholine and serotonin (5-hydroxytryptamine [5-HT]) contributes to dysmotility in GERD.Citation9
5-HT receptor 4 (5-HT4) agonists stimulate the release of acetylcholine at the myenteric plexus, leading to increased gastrointestinal contractions.Citation10 Revexepride is a highly selective 5-HT4 agonist, and is a member of a new class of prokinetic agents that stimulate gastric emptying and that have potential as a treatment for GERD.Citation11 Revexepride was developed as a potential add-on therapy to PPIs specifically for patients who have persistent symptoms of regurgitation, with or without heartburn, while on PPI therapy.Citation5,Citation12,Citation13 However, two Phase II trials (ClinicalTrials.gov Identifiers: NCT01472939 and NCT01370863) showed no consistent benefit of revexepride over placebo in terms of symptom or reflux events.Citation5,Citation12
The aim of this Phase I study was to investigate the pharmacokinetics and excretion of a single oral dose of 14C-labeled revexepride in healthy volunteers using a microtracer approach with accelerator mass spectrometry (AMS). In the last decade, the administration of reduced doses of 14C-labeled compounds (typically 0.2–1 µCi) combined with AMS has enabled quantitative absorption, metabolism, and excretion (AME) studies to be conducted in place of traditional scintillation counting-based studies.Citation14 The very low doses of radioactivity used in these studies minimize potential safety and regulatory concerns around the use of radioactive material in clinical trials.Citation14 Absorption and excretion results, as well as safety and tolerability data, are presented in this study. Metabolite data are reported elsewhere.Citation15
Participants and methods
Study design
This was an open-label, non-randomized, AME study (ClinicalTrials.gov Identifier: NCT01786876; ). The metabolism methodology and results are published elsewhere.Citation15 Screening took place in the 28 days preceding day 1, and included demographics, vital signs, electrocardiograms (ECGs), drug and alcohol screening, medical history, and clinical laboratory assessments. On day 1, following an overnight fast of at least 8 hours (water was permitted), all participants received a single oral capsule of 2 mg [14C]revexepride free-base equivalent (as monohydrochloride monohydrate salt) containing ~200 nCi of radioactivity. Radiolabeled material was manufactured and provided by Johnson & Johnson (Movetis NV, now part of the Shire group of companies). Samples for safety and pharmacokinetic data were collected on each of days 1–10, and at clinic discharge on day 11. Follow-up telephone calls took place 7±2 days after discharge. Revexepride concentrations were determined in plasma and urine by liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS). Total 14C radioactivity was determined for blood, plasma, urine, and fecal samples by AMS.
Figure 1 Study flow and timeline.
The study was carried out in accordance with the International Conference on Harmonisation of Good Clinical Practice, the principles of the Declaration of Helsinki, and all applicable local ethical and legal requirements. The study protocol was reviewed by the relevant institutional review board (Schulman Associates IRB, FL, USA). Before entering the study, all participants provided written informed consent.
Study participants, and inclusion and exclusion criteria
Study participants (men aged 18–50 years, inclusive) were eligible for inclusion if they had a body mass index of 18.5–30.0 kg/m2, and no clinically relevant diseases that could affect the pharmacokinetics of revexepride, as determined by the screening assessments. Participants were also required either to be sterile or to use, from day −1 until 90 days following clinic discharge, an approved method of contraception. In addition, participants were asked to refrain from strenuous exercise (from 48 hours before day −1 until the follow-up telephone call). Exclusion criteria were the following: cholecystectomy and/or appendectomy; history of or current marked prolongation of the QT/corrected QT interval (>450 ms); current or relevant past history of physical or psychiatric illness, any medical disorder that might require treatment or make the participant unlikely to complete the study, or any condition that meant the participant would be at undue risk from the investigational product or study procedures; current use of any medication (including over-the-counter, herbal, or homeopathic preparations) except the occasional use of paracetamol (acetaminophen); known or suspected intolerance to the investigational product or to any excipients; previous exposure to HIV or to hepatitis B or C virus; consumption of >21 units of alcohol per week or >3 units per day; use of tobacco in any form; other substance abuse within the past year; more than two bowel movements per day, fewer than three bowel movements per week, or a significant change in bowel habits in the 30 days before receiving the first dose of investigational product; participation in a study involving ingestion of 14C in the 6 months preceding the study; or exposure to clinically significant radiation in the 12 months before the study.
Study end points
The study end points were mass balance and pharmacokinetic parameters. Pharmacokinetic parameters were determined from the plasma and blood concentration–time data of total radioactivity and the plasma concentration–time data for revexepride by non-compartmental analysis. Parameters calculated for blood and plasma included (but were not limited to) maximum concentration (Cmax), time to Cmax (Tmax), area under the concentration–time curve from time 0 to the last measurable concentration (AUC0−t) and extrapolated to infinity (AUC0−∞), disposition half-life (t1/2), clearance from plasma after oral administration (CL/F), terminal elimination rate constant (λz), and volume of distribution (Vz/F; revexepride in plasma only). Urine total radioactivity and revexepride concentrations were used to calculate the total radioactivity excreted in urine (Aeu), the proportion of radioactivity excreted, and renal clearance (CLR). Fecal total radioactivity concentrations were used to calculate the total radioactivity excreted in feces (Aef) and the proportion excreted.
Sample collections
Serial blood samples were collected prior to drug administration and at 0.5, 1, 1.5, 2, 2.5, 4, 6, 8, 12, and 24 hours post-dose and then at 24-hour intervals until day 11 (240 hours post-dose). Blood samples were drawn into potassium ethylenediamine tetraacetic acid tubes, sealed, mixed by inversion, and chilled immediately on ice. An aliquot of whole blood was removed, and the remainder of each sample was centrifuged to prepare plasma. Urine and stool samples were collected prior to (day −1) and after dosing (day 1) and for a further 10 days. Individual urine and stool samples were stored and subsequently pooled by collection period. Urine was pooled at 0–2, 2–4, 4–8, 8–12, and 12–24 hours, and then at 24-hour intervals until day 11 (240 hours post-dose). Feces were pooled across 24-hour intervals until 240 hours post-dose. Pooled fecal samples were homogenized with water (1:1), and a subsample (~20 g) was lyophilized (stored at 4°C) and then ground to a fine powder. Blood, urine, and fecal samples were stored at −80°C.
Bioanalytical methodologies
Liquid chromatography–mass spectrometry/mass spectrometry
Plasma and urine samples were assayed to determine revexepride-base concentrations using previously validated LC–MS/MS methodology (Analytical Biochemical Laboratory BV, Assen, the Netherlands). For plasma samples, this method was linear over the range 2–1,000 pg/mL with a lower limit of quantification (LLOQ) of 2 pg/mL. For urine samples, the LLOQ was 1 ng/mL, and the calibration curve ranged from 1 to 1,000 ng/mL for revexepride base. Quality control samples were either prepared using human plasma or human urine controls at three concentration levels. The quality control samples were stored with and assayed alongside each batch of study samples, with freshly prepared calibration standards. Quality control and calibration standard data were accepted in accordance with the Food and Drug Administration guidelines.Citation16
Accelerator mass spectrometry
Radioactivity levels of 14C in plasma, whole blood, and urine were analyzed by AMS (single-stage accelerator mass spectrometer-250; NEC, Tokyo, Japan). Urine samples were diluted before analysis to minimize the risk of urea impacting the graphitization process. The appropriate diluent was determined by diluting pre-dose urine with water or buffer at varying levels (tenfold, 100-fold, 500-fold, and 1,000-fold) followed by addition of [14C]revexepride to each prepared dilution at the same concentration. The resulting 14C concentration data obtained from these dilutions were compared with those from water or buffer containing an equivalent concentration of [14C]revexepride. The dilution factor and diluent of the diluted urine sample that most accurately reflected the concentration measured in the spiked buffer or water (a tenfold dilution with water in this instance) were selected and used to dilute all pre-dose and post-dose urine samples prior to analysis.
Plasma, whole blood, urine, and freeze-dried fecal samples and controls were placed in clean, glass sample tubes containing prebaked copper oxide powder, and the mixture was dried under vacuum. For samples containing minimal amounts of carbon, that is, diluted urine, carbon carrier was added in the form of sodium benzoate. A single analysis was performed per sample, with the exception of freeze-dried fecal samples, which were analyzed in duplicate. Oxalic acid (equivalent to 5–7 mg), placed in separate sample tubes containing prebaked copper oxide powder, and sample tubes containing a defined amount of sodium benzoate carbon carrier and prebaked copper oxide were also dispensed as standards. All standards and controls were dried under vacuum. The final amount of carbon used for graphitization was ~2 mg. The samples and controls were graphitized according to previously published methods.Citation17
The AMS data were used to calculate total radioactivity for each sample type and the percentage of the administered radioactivity dose excreted in feces and in urine. The 14C concentrations measured in the pre-dose samples of each sample type were used for background subtraction.
Safety measurements
Adverse event information was collected from the time of signing of the informed consent form until follow-up telephone calls 7 days after discharge. Blood pressure and pulse rate, respiratory rate, and oral temperature were measured during all study stages (), except the follow-up call period. A complete physical examination was performed by a qualified physician on day −1 and at discharge. A 12-lead ECG was conducted during all study stages (), except the follow-up call period. Cotinine (to monitor for tobacco use) and drug and alcohol screens were conducted on day −1. Serology tests were conducted during screening.
Clinical laboratory measurements
Clinical laboratory measurements were performed during screening, on day −1, and at discharge/early termination. Measurements included biological, hematological, and urinalysis parameters.
Statistical analyses
Statistical analyses were performed using SAS® Version 9.1.3 (or newer versions) (SAS Institute Inc., Cary, NC, USA). Summary statistics (number of observations, mean, standard deviation [SD], coefficient of variation, median, maximum, minimum, and geometric mean) were determined for all pharmacokinetic parameters. Plasma concentrations at each nominal sampling time were also summarized using descriptive statistics.
Results
Study participants
Six healthy adult men were enrolled in the study. Their mean age was 29.5 years (SD, 2.07 years), and their mean body mass index was 25.0 kg/m2 (SD, 2.11 kg/m2; ). All six participants completed the study and were included in the analysis.
Table 1 Study demographics and participant characteristics
Mass balance and pharmacokinetic parameters
By 240 hours post-dose, a mean total of 38.2% (SD, 10.3%) of radioactivity had been excreted in urine (range, 27.8%–50.6%), and a mean total of 57.3% (SD, 10.4%) in feces (range, 45.3%–69.6%; and ), giving a mean total recovery of 95.5% (SD, 4.5%; range, 87.1%–99.8%).
Table 2 Mean data for urinary and fecal 14C dose excretion
Figure 2 Mean cumulative percent of radioactive dose recovered in urine and feces following administration of a single dose of 2 mg [14C]revexepride to healthy male volunteers in the pharmacokinetic analysis set (linear scale).
![Figure 2 Mean cumulative percent of radioactive dose recovered in urine and feces following administration of a single dose of 2 mg [14C]revexepride to healthy male volunteers in the pharmacokinetic analysis set (linear scale).](/cms/asset/78fb43e0-c918-4059-870b-0c4c73bc2a27/dddt_a_12166217_f0002_b.jpg)
Plasma total radioactivity concentrations were quantifiable up to 24 (four participants) and 48 hours (two participants) post-dose. Whole-blood total radioactivity concentrations were quantifiable up to 12 (three participants) and 24 hours (three participants) post-dose. Plasma concentrations of revexepride were quantifiable up to 72 (one participant), 96 (two participants), 120 (two participants), and 240 hours (one participant) post-dose ().
Figure 3 Concentration of total radioactivity in blood and plasma and revexepride in plasma.
Abbreviation: LC–MS/MS, liquid chromatography–mass spectrometry/mass spectrometry.
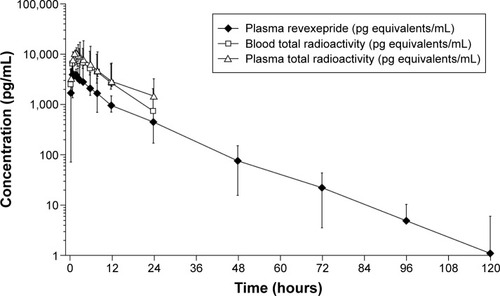
Pharmacokinetic parameter data are shown in . Mean (± SD) values of Tmax for radioactivity in plasma, radioactivity in whole blood, and revexepride in plasma indicated rapid absorption (1.83±1.13, 2.17±1.03, and 1.75±1.17 hours, respectively). Mean (± SD) values of t1/2 for radioactivity in plasma, radioactivity in whole blood, and revexepride in plasma were comparable (10.9±2.54, 6.96±1.51, and 11.0±2.38 hours, respectively). After Tmax, plasma concentrations of revexepride and concentrations of total radioactivity in plasma and whole blood appeared to decline in a biphasic manner. The blood plasma radioactivity ratio was ~0.8 (range, 0.72–0.92); revexepride Cmax and AUC accounted for 30%–40% of the total radioactivity exposure in plasma. The total mean ± SD plasma clearance (CL/F) of revexepride was 49.8±15.2 L/h, and the mean ± SD Vz/F of revexepride was 828±118 L. The apparent mean CLR of revexepride was 8.6 L/h (or 143 mL/min). The mean urinary excretion of revexepride accounted for 18.3% of the administered dose.
Table 3 Pharmacokinetic parameter data in whole blood and plasma
The geometric mean plasma AUC(0−∞) of revexepride accounted for 32.0% of the total plasma radioactivity.
Adverse events and other safety results
All six participants had at least one treatment-emergent adverse event (TEAE) during the study. Five participants had seven TEAEs of diarrhea, all of which were considered related to treatment. One participant experienced a physical procedural complication (scratch), but this was not considered to be related to the study drug. All TEAEs were mild in severity. There were no serious TEAEs, and no TEAEs led to study discontinuation. All ECGs were considered normal, with no shifts to abnormal observed. No clinically meaningful changes in vital signs, ECG parameters, or clinical laboratory measurements were observed.
Discussion
This study presents pharmacokinetic and excretion data for [14C]revexepride. The overall recovery of radioactivity was very high, with a mean recovery of almost 100% of the administered 14C dose. This is a substantial amount given that, in general, total radioactivity recovered in this type of study is <90%.Citation18
Urinary recovery was moderate (~38%), indicating moderate-to-good oral absorption. Fecal recovery levels of 14C were higher (nearly 60% of total recovery) than urinary recovery levels, which may indicate unabsorbed material or biliary-excreted unchanged drug or metabolites. The blood plasma ratio of 0.8 for total radioactivity suggests a low uptake into blood cells. Revexepride Cmax and AUC accounted for 30%–40% of the total radioactivity exposure in plasma, which indicated that 60%–70% of radioactivity was from metabolites. The geometric mean plasma AUC of revexepride accounted for 32.0% of the total plasma radioactivity exposure, indicating the presence of circulating metabolites. The mean plasma and blood total radioactivity fell below the LLOQ for a number of participants at later time points; these values were reported as zeros for the calculation of summary statistics (). It should be noted that the LLOQ for LC–MS/MS was lower than that of the assays used to determine total radioactivity, allowing plasma revexepride to be monitored for a longer period of time following administration ().
The apparent mean CLR for revexepride was 8.6 L/h (or 143 mL/min). This is slightly higher than the typical glomerular filtration rate in healthy individuals of a similar mean age (28.2 years) to those in the current study,Citation19 suggesting some contribution of active secretion in the renal tubules by one or more transporters. Revexepride was rapidly absorbed, with Cmax reached in ~2 hours, and the observed t1/2 was ~11 hours, indicating good drug stability in plasma.
The pharmacokinetic parameters of Cmax, Tmax, and t1/2 in the current study were similar to those reported in a previous study of revexepride in healthy volunteers.Citation13 However, the AUC was approximately double the AUC observed in the earlier study, which is likely a reflection of the higher dose used in the current study (2 vs 1 mg).
All six participants experienced diarrhea following revexepride administration, which is an expected adverse event of prokinetic drugs, especially as the participants were healthy with no constipation. Prokinetic agents stimulate gastric motility and emptying; diarrhea in healthy volunteers is therefore not an unexpected side effect and has been reported for other prokinetic agents.Citation20–Citation22 No serious TEAEs occurred, and no TEAEs led to study discontinuation. This study was not specifically designed to monitor the development of cardiovascular TEAEs; however, it is worth noting that no cardiovascular adverse events or clinically meaningful changes in ECGs were seen during the study. Cardiovascular adverse events have been of concern with nonselective 5-HT4 agonists, such as cisapride (QT prolongation) and tegaserod (ischemia), leading to the withdrawal and restricted use of these drugs.Citation23 These adverse effects have been suggested to involve off-target interactions with the hERG-encoded potassium channel or the 5-HT1 and 5-HT2 receptors expressed in the cardiovascular system.Citation23 However, the newer, selective 5-HT4 agonists are not believed to be associated with adverse cardiovascular effects.Citation23,Citation24 Consistent with this, other clinical trials involving revexepride reported no cardiac concerns.Citation5,Citation12,Citation13
This study utilized a microtracer approach combined with AMS; this type of approach exhibits a number of strengths over conventional liquid scintillation-based methodologies.Citation14 First, the increased sensitivity of AMS allows lower doses of radiolabeled compounds to be administered, reducing the need to perform preclinical AME studies in animals.Citation25 Second, these studies avoid problems associated with the labeled material being radiochemically unstable at high specific radioactivity and prone to radiolysis, or with study participants being limited in the radioactive dose that they can receive safely.Citation26,Citation27 Finally, a small amount of radioactivity is combined with doses of clinical relevance, thereby overcoming issues relating to potential dosing nonlinearity.Citation28,Citation29
Conclusion
This study demonstrated that revexepride was rapidly absorbed and exhibited an appreciable level of oral absorption. Almost 100% of total radioactivity was recovered in the urine (mean, 38.2%) and feces (mean, 57.3%) of participants, indicating that these were the major routes of excretion. No safety signals were identified. The clinical development of revexepride for GERD has now been halted due to the results of the Phase II trials;Citation5,Citation12 however, these data may be important for future clinical studies.
Acknowledgments
Robert Gillies, PhD, and Luci Witcomb, PhD, from PharmaGenesis, London, UK, provided editorial assistance for this manuscript, funded by Shire. The authors would also like to thank Lynne Poole and Patrick Martin of Shire for their assistance in preparing the manuscript, and the study participants for their involvement in the study.
Disclosure
The study was funded by Shire International GmbH, Eysins, Switzerland. Covance Laboratories Inc. was funded by Shire to conduct the clinical trial. Xceleron Inc was funded by Shire to perform AMS and LC–MS/MS analysis. PharmaGenesis London, UK, was funded by Shire to provide editorial assistance with the preparation of manuscript. Steven Troy and Jay Getsy are employees of Shire and also hold stocks/share options in Shire and report no other conflicts of interest in this work. Ron Budhram, Mike Pennick, and Graeme Scarfe are former employees of Shire and report no other conflicts of interest in this work. Stephen Flach, Marie Croft, Jie Ding, and Todd Pankratz report no conflicts of interest.
References
- El-SeragHBSweetSWinchesterCCDentJUpdate on the epidemiology of gastro-oesophageal reflux disease: a systematic reviewGut201463687188023853213
- VakilNvan ZantenSVKahrilasPDentJJonesRThe Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensusAm J Gastroenterol2006101819001920 quiz 194316928254
- KatzPOTutuianRHistamine receptor antagonists, proton pump inhibitors and their combination in the treatment of gastro-oesophageal reflux diseaseBest Pract Res Clin Gastroenterol200115337138411403533
- FassRGasiorowskaARefractory GERD: what is it?Curr Gastroenterol Rep200810325225718625135
- ShaheenNJAdlerJDedrieSRandomised clinical trial: the 5-HT4 agonist revexepride in patients with gastro-oesophageal reflux disease who have persistent symptoms despite PPI therapyAliment Pharmacol Ther201541764966125693609
- VelaMFCamacho-LobatoLSrinivasanRTutuianRKatzPOCastellDOSimultaneous intraesophageal impedance and pH measurement of acid and nonacid gastroesophageal reflux: effect of omeprazoleGastroenterology200112071599160611375942
- HershcoviciTFassRManagement of gastroesophageal reflux disease that does not respond well to proton pump inhibitorsCurr Opin Gastroenterol201026436737820571388
- ChampionMCProkinetic therapy in gastroesophageal reflux diseaseCan J Gastroenterol199711Suppl B55B65B
- SaegusaYTakedaHMutoSDecreased motility of the lower esophageal sphincter in a rat model of gastroesophageal reflux disease may be mediated by reductions of serotonin and acetylcholine signalingBiol Pharm Bull201134570471121532161
- De MaeyerJHLefebvreRASchuurkesJAJ5-HT4 receptor agonists: similar but not the sameNeurogastroenterol Motil20082029911218199093
- Bruley des VarannesSCoronEGalmicheJPShort and long-term PPI treatment for GERD. Do we need more-potent anti-secretory drugs?Best Pract Res Clin Gastroenterol201024690592121126703
- TackJZerbibFBlondeauKRandomized clinical trial: effect of the 5-HT4 receptor agonist revexepride on reflux parameters in patients with persistent reflux symptoms despite PPI treatmentNeurogastroenterol Motil201527225826825530111
- PierceDCorcoranMVelinovaMHossackSHoppenbrouwersMMartinPA phase 1 randomized study evaluating the effect of omeprazole on the pharmacokinetics of a novel 5-hydroxytryptamine receptor 4 agonist, revexepride (SSP-002358), in healthy adultsDrug Des Devel Ther2015912571268
- HahSSHendersonPTTurteltaubKWRecent advances in biomedical applications of accelerator mass spectrometryJ Biomed Sci2009161111
- BobbaSDingJChowdhuryGUtilization of AMS and HRMS for metabolite profiling and identification of [14C]Revexepride (SSP-002358) in a human microtracer phase 1 study62nd ASMS Conference on Mass Spectrometry and Allied TopicsJune 15–19, 2014Baltimore, MD
- Food and Drug Administration (FDA). Center for Drug Evaluation and Research (CDER). Guidance for industryBioanalytical method validation2001 Available from: http://www.fda.gov/downloads/Drugs/.../Guidances/ucm070107.pdfAccessed January 26, 2016
- VogelJSRapid production of graphite without contamination for biomedical AMSRadiocarbon1992343344350
- RoffeySJObachRSGedgeJISmithDAWhat is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabeled drugsDrug Metab Rev2007391174317364879
- VervoortGWillemsHLWetzelsJFAssessment of glomerular filtration rate in healthy subjects and normoalbuminuric diabetic patients: validity of a new (MDRD) prediction equationNephrol Dial Transplant200217111909191312401845
- De SchryverAMAndriesseGISamsomMSmoutAJGooszenHGAkkermansLMThe effects of the specific 5HT4 receptor agonist, prucalopride, on colonic motility in healthy volunteersAliment Pharmacol Ther200216360361211876716
- CamilleriMVazquez-RoqueMIBurtonDPharmacodynamic effects of a novel prokinetic 5-HT receptor agonist, ATI-7505, in humansNeurogastroenterol Motil2007191303817187586
- TackJVosRJanssensJSalterJJauffretSVandeplasscheGInfluence of tegaserod on proximal gastric tone and on the perception of gastric distensionAliment Pharmacol Ther200318101031103714616170
- TackJCamilleriMChangLSystematic review: cardiovascular safety profile of 5-HT(4) agonists developed for gastrointestinal disordersAliment Pharmacol Ther201235774576722356640
- MendzelevskiBAusmaJChanterDOAssessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT studyBr J Clin Pharmacol201273220320921848574
- CombesRDBerridgeTConnellyJEarly microdose drug studies in human volunteers can minimise animal testing: proceedings of a workshop organised by Volunteers in Research and TestingEur J Pharm Sci200319111112729856
- ArjomandAAccelerator mass spectrometry-enabled studies: current status and future prospectsBioanalysis20102351954120440378
- BeumerJHGarnerRCCohenMBHuman mass balance study of the novel anticancer agent ixabepilone using accelerator mass spectrometryInvest New Drugs200725432733417347871
- GianniLKearnsCMGianiANonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humansJ Clin Oncol19951311801907799018
- KaulSShuklaUABarbhaiyaRHNonlinear pharmacokinetics of nefazodone after escalating single and multiple oral dosesJ Clin Pharmacol19953588308398522641