
Abstract
Neuropathy is a serious complication of diabetes that has a significant socioeconomic impact, since it frequently demands high levels of health care consumption and compromises labor productivity. Recently, LASSBio-1471 (3) was demonstrated to improve oral glucose tolerance, reduce blood glucose levels, and display an anti-neuropathy effect in a murine streptozotocin-induced diabetes model. In the present work, we describe the design, synthesis, solubility, plasma stability, and pharmacological evaluation of novel sulfonylhydrazone derivatives (referred to herein as compounds 4–9), which were designed by molecular modification based on the structure of the prototype LASSBio-1471 (3). Among the compounds tested, better plasma stability was observed with 4, 5, and 9 in comparison to compounds 6, 7, and 8. LASSBio-1773 (7), promoted not only hypoglycemic activity but also the reduction of thermal hyperalgesia and mechanical allodynia in a murine model of streptozotocin-induced diabetic neuropathic pain.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
Neuropathy is a serious complication of diabetes that has a significant socioeconomic impact, since it frequently demands continued health care and compromises labor productivity.Citation1–Citation5 Diabetic peripheral neuropathic pain is currently treated based on glycemic control and pain management.Citation6–Citation8 Several prototypes and drugs under development are being studied for treatment of diabetic peripheral neuropathic pain.Citation9 Recently, Freitag and Miller demonstrated that PPAR agonists could be effective in the treatment of various animal and human pain conditions.Citation10,Citation11 Rosiglitazone (1) and pioglitazone (2) are synthetic PPARγ agonists currently used to treat type 2 diabetes mellitus. However, safety concerns including increased plasma creatinine and homocysteine, signs of myopathy and rhabdomyolysis, fluid retention, peripheral edema, weight gain, and a potential increased risk of cardiac failure, bring into question the safety of these drugs.Citation12 Zapata-Sudo et al identified new synthetic PPARγ agonists, described as sulfonylhydrazone derivatives; long-term administration (20 mg/kg, intraperitoneally [ip], for 7 days) of compound 3′-[(E)-[2-[(6-methyl-1,3-benzodioxol-5-yl)sulfonyl] hydrazinylidene]methyl]benzoic acid (3, LASSBio-1471) improved oral glucose tolerance, reduced blood glucose levels, and reduced neuropathic pain in a murine streptozotocin (STZ)-induced diabetes model.Citation13
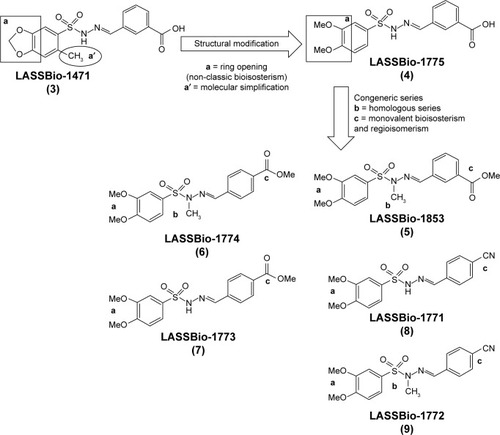
This work describes the design, synthesis, solubility, plasma stability, and pharmacological evaluation of novel sulfonylhydrazones (referred to herein as compounds 4–9), designed by molecular modification based on the structure of prototype LASSBio-1471 (3) ().
Figure 1 Genesis concept of the novel sulfonylhydrazones 4–9 from molecular modifications based on prototype 3.
Results and discussion
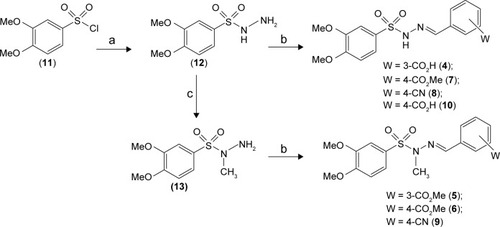
Compounds 4–9 were synthetized as depicted in . Sulfonylhydrazones 4, 7, and 8 were prepared in two linear steps based on hydrazinolysis of starting material 11 followed by condensation with functionalized aldehydes to produce good yields of the target compounds.Citation14 N-methyl-sulfonylhydrazone analogs (5, 6, and 9) were synthetized in a similar manner using N-methyl-sulfonylhydrazide 13 as a key intermediate. This compound was obtained with 39.5% overall yield through protection from phthalic anhydride, followed by N-methylation with methyl iodide and deprotection of phthalimide, using hydrazine hydrate in ethanol ().Citation15,Citation16 The 1H and 13C nuclear magnetic resonance (NMR) spectral analysis of compounds 4–9 revealed the presence of only one signal related to imine hydrogen (N=CH), indicating that compounds were synthesized as a single isomer. The assignment of the possible geometrical isomer obtained (ie, Z or E) was performed considering the chemical shift of the imine hydrogen and based on previous data from literature.Citation13–Citation17 These data indicated that compounds 4–9 were obtained as E-diastereoisomers (N=CH in configuration E).
Figure 2 Reagents and conditions.
In order to obtain information about the druglikeness profile of compounds 4–9, their cLogP, and tPSA were calculated in silico and their aqueous solubility and pKa values were determined using ultraviolet spectrometry.Citation18,Citation19 The data summarized in showed that compounds 4–9 have ideal cLogP values and satisfactory to good aqueous solubility, with the exception of compound 6 (LASSBio-1774).
Table 1 Physicochemical properties and aqueous solubility of compounds 4–9
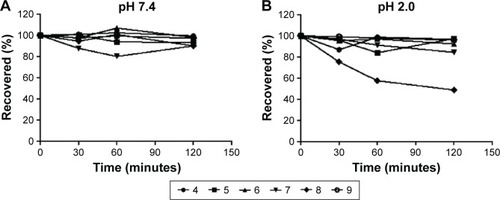
The chemical stability of sulfonylhydrazones 4–9 in buffer solution with pH values that simulate gastric acid (pH =2.0) and serum content (pH 7.4) was investigated.Citation20 As shown in , all compounds presented great chemical stability, with the exception of compound 8 that was unstable at pH 2.0.
Figure 3 In vitro chemical stability.
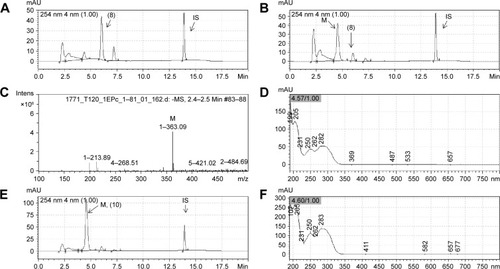
Compounds 4–9 were also studied in order to determine their plasma stability.Citation20,Citation21 After 60 minutes incubation with rat plasma, compounds 4, 5, and 9 were very stable, while compounds 6, 7, and 8 were unstable (). Only 2.2% and 9.6% of compounds 6 and 7, respectively, were recovered after plasma incubation, indicating the high lability of the ester subunit. A mass chromatogram was obtained from the analysis of compounds 7 and 8 and their metabolites, generated using rat plasma, by high performance liquid chromatography/mass spectrometry (HPLC/MS) ( and ). As shown in , the metabolite M of compound 7 (LASSBio-1773) appears at retention time 4.57 and displays a peak with m/z value of 363.04 in negative mode suggestive of metabolite resulting from the loss of 15 atomic units, indicating the possibility of the hydrolysis of carboxymethyl ester moiety, presented in the structure of compound 7 (m/z 378.04). In order to unequivocally identify the plasma metabolite of compound 7, carboxylic acid derivative 10 was synthetized () and co-injected for 60 minutes incubation with rat plasma samples. The ultraviolet spectroscopy (UV) analysis of synthesized metabolite 10 presented a great correlation with the rat plasma metabolite M (), and a complete superposition between the peaks of metabolite M and the standard 10 was observed (). Considering that the cyano group, presented in compound 8, can also be hydrolyzed to the corresponding carboxylic acid, the same methodology was applied for the analysis of its metabolite (). As shown in , the plasma metabolite of compound 8 (m/z 345.09) appears at the same retention time (4.57) as the metabolite identified for compound 7, and displayed a peak with an m/z value of 363.09. These results were suggestive of hydrolysis of the cyano subunit. To confirm this hypothesis, carboxylic acid derivative 10 was co-injected for 60 minutes incubation with rat plasma samples previously treated with compound 8. The UV analysis of the standard 10 and the plasma metabolite of compound 8 presented higher correlation and great overlap between the peaks of metabolite M and the standard 10 ().
Table 2 In vitro stability of compounds 4–9 in rat plasma
Figure 4 Representative chromatograms of LASSBio-1773 (7) (20 µM) and its metabolite formed by incubation with rat plasma.
Abbreviations: HPLC/MS, high performance liquid chromatography/mass spectrometry; min, minutes; Intens, Intensity.
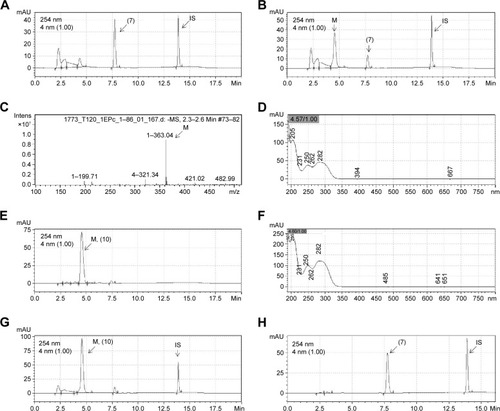
Figure 5 Representative chromatograms of LASSBio-1771 (8) (20 µM) and its metabolite formed by incubation with rat plasma.
Abbreviations: HPLC/MS, high performance liquid chromatography/mass spectrometry; min, minutes; UV, ultraviolet spectroscopy; Intens, Intensity.
The anti-diabetic profile of compounds 4–9 was investigated through the evaluation of their ability to reduce blood glucose levels in a murine model of diabetes induced by STZ.Citation13,Citation14 Among these compounds, only sulfonylhydrazones 6 and 7 showed hypoglycemic activity. After ip administration of a single dose, LASSBio-1773 (20 mg·kg−1) (7) significantly reduced blood glucose from 384.0±41.9 mg/dL to 182.2±22.7 mg/dL and LASSBio-1774 (6) reduced it from 397.1±38.2 mg/dL to 255.3±42.2 mg/dL ().
Figure 6 Activity of compounds 4–9 in a murine model of diabetes induced by streptozotocin.
Abbreviation: DMSO, dimethyl sulfoxide.
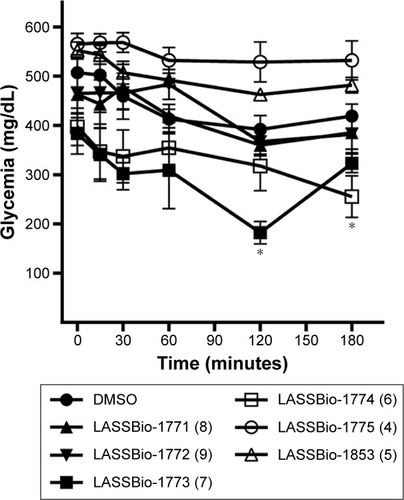
Compound 9 (LASSBio-1772) did not reduce hyperglycemia after treatment with a single dose (), although it was recently described by Lima et al as a partial agonist of PPARγ.Citation14 When administered for 7 days at a dose of 50 mg/kg, it produced a significant reduction in glucose levels (242.4±55.3 mg/dL) when compared to vehicle (410.0±58.0 mg/dL).
Additionally, we investigated the anti-diabetic profile of sulfonylhydrazone 7 (LASSBio-1773) during administration of 50 mg/kg once a day for 7 days. The effect on blood glucose levels was measured before and 3 and 7 days after treatment with LASSBio-1773 (7). As shown in , compound 7 promoted hypoglycemic activity only after 7 days of treatment.
Table 3 Evaluation of blood glucose levels (mg/dL) in diabetic rats treated with vehicle (DMSO) and LASSBio-1773 (7), 50 mg·kg−1, ip for 7 days
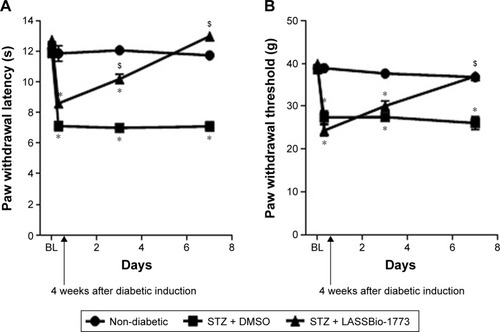
Considering the fact that the sulfonylhydrazone 9 (LASSBio-1772), which has high chemical and plasma stability ( and ), had been described as an antinociceptive agent in a diabetic neuropathic pain model induced by STZ in rats,Citation14 analog 7 (LASSBio-1773) was selected to be evaluated on the same experimental model. As shown in , long-term administration of compound 7 (dose 50 mg·kg−1, ip) reduced diabetic neuropathic pain. Four weeks after induction of diabetes, the paw withdrawal latency (PWL, ) and paw withdrawal threshold (PWT, ) were significantly reduced from 13.0±0.4 s to 8.5±0.2 s and from 39.8±0.2 g to 24.0±1.4 g, respectively, showing the induced diabetic neuropathy. Compound 7 (LASSBio-1773) significantly increased the PWL from 8.5±0.2 seconds to 12.2±0.9 seconds and 13.3±0.2 seconds, after 3 and 7 days of treatment, respectively (). It also increased the PWT from 24.0±1.4 g to 36.7±0.9 g ().
Figure 7 Effect of administration of compound 7 (LASSBio-1773) on diabetic neuropathy.
Abbreviations: DMSO, dimethyl sulfoxide; STZ, streptozotocin; s, seconds.
In order to investigate the mechanism of the action of compounds 4–9 and considering that these compounds were designed based on molecular modification of the prototype LASSBio-1471 (3), previously described as PPARγ ligand,Citation14 PPARγ binding assay was performed by CEREP®, employing a single concentration of compounds 4–9 (C =30 µM) using rosiglitazone as positive control. In this model, these compounds were not able to inhibit the control specific binding to PPARγ. In a similar manner, compound 10, identified as the plasma metabolite of 7 and 8, was also evaluated and was unable to bind to PPARγ (data not shown).
In summary, a new series of sulfonylhydrazones (4–9) was synthetized, and their druglikeness profile (including cLogP, aqueous solubility, and pKa), and chemical and plasma stability were determined. From this series, compounds 4, 5, and 9 showed the best plasma stability, while compounds 6, 7, and 8 presented high instability in rat plasma. Considering their pharmacological profile, LASSBio-1773 (7) has displayed expressive hypoglycemic activity, by intraperitoneal administration, in a diabetes model induced by STZ. It was also able to reduce the thermal hyperalgesia and mechanical allodynia in a murine model of diabetic neuropathic pain induced by STZ. These activities seem not to be related to the PPARγ, since these compounds were not able to bind to these receptors. The mechanism of action of these compounds is now under investigation.
Experimental pharmacology
All experiments were approved by the Ethics Committee on Animal Use in Scientific Experimentation Center of Health Sciences, Federal University of Rio de Janeiro which follows the guidelines and is registered with the National Board of Animal Experimentation Control. Animals were housed three per cage at constant temperature of 23°C±2°C under 12-hour light/dark cycle, with food and water ad libitum.
The compounds were tested in diabetic rats (Rattus norvegicus). Experimental diabetes was induced through a single intravenous injection of STZ (60 mg·kg−1; Sigma-Aldrich Co., St Louis, MO, USA) dissolved in citrate buffer (pH 4.5). After 4 weeks, male Wistar rats (R. norvegicus weighing 180–220 g) with glycemia >300 mg/dL were considered diabetic. Animals were randomly divided into three groups: non-diabetic, diabetic treated with solvent polyethylene glycol (dimethyl sulfoxide [DMSO], Merck Millipore, Billerica, MA, USA), and diabetic treated with compounds. Compounds 4–9 were administered at a single dose (20 mg·kg−1, ip) and plasma glucose levels were measured at 0, 30, 60, 120, and 180 minutes after treatment, using commercial kit Accu-Chek® Performa Monitoring System (Hoffman-La Roche Ltd., Basel, Switzerland). The anti-diabetic profile was also observed during long-term administration of compound 7 at a dose of 50 mg/kg, ip.
Nociceptive test
Thermal hyperalgesia and mechanical allodynia
Thermal hyperalgesia was determined by measuring PWL assessed by applying radiant heat to the hind paw using a plantar analgesia meter (model 33ITC Inc., CA, USA). Mechanical allodynia was determined by measuring PWT to pressure applied to the hind paw using a digital analgesiometer (model EFF301, Insight, São Paulo, Brazil). PWL, PWT, and plasma glucose were determined before and after 4 weeks of diabetes induction, as well as 3 and 7 days after treatment with compound 7 (LASSBio-1773) at a dose of 50 mg·kg−1 ip.
Statistical analysis
Values are expressed as mean ± standard error of the mean and analyzed using GraphPrism software (version 5.0). One-way analysis of variance (ANOVA) test followed by Dunnett post-tests or ANOVA two-way test were used for comparisons among groups, and were considered significant for all tests with a minimum of P<0.05.
Chemical – HPLC/UV
In this study, we reported the synthesis of new sulfonylhydrazone derivatives (4–9). Structural characterizations of the synthesized compounds were made by using spectroscopic methods, magnetic measurements, and thermal studies. Melting points were determined using Quimis 340 apparatus and are uncorrected. 1H NMR spectra were determined in deuterated chloroform or DMSO containing approximately 1% tetramethylsilane as an internal standard, using Bruker DPX-200 at 200 MHz. 13C NMR spectra were determined with the same spectrometer at 50 MHz, using the same solvents. The progress of all reactions was monitored through thin layer chromatography (HF-254, Merck Millipore). The developed chromatograms were viewed under ultraviolet light (254–265 nm) and treated with iodine vapor. Reagents and solvents were purchased from commercial suppliers and used as received.
Purity of compounds 4–9 and metabolite 10 was determined through HPLC (>95%) using Shimadzu – LC20AD apparatus, Kromasil 100-5C18 (4.6 mm×6,250 mm) column, and SPD-M20A detector (Diode Array) at 254 nm for quantification of analyte in a 1 mL/min constant flux. The injector was programmed to inject a volume of 20 µL. The solvent systems used were: CH3CN:H2O 50:50 and 60:40, and isocratic HPLC mode was used.
Ultraviolet spectroscopy was performed using a Femto spectrophotometer. The wavelength used in the solubility assay was determined by the λmax characteristic of each compound. Spectra were analyzed with Femtoscan software.
Procedure to prepare 3,4-dimethoxybenzenesulfonohydrazide (12)
A solution of the appropriate sulfonyl chloride (11) (4.24 mmol) at 0°C for 5 minutes was slowly added to a stirred solution of hydrazine hydrate (16.95 mmol) in CHCl3 (20 mL). The reaction mixture was stirred at room temperature for 4 hours until the end of the reaction. Subsequently, 30 mL water saturated with NaCl was added. The layers were separated, and the aqueous phase was extracted with CH2Cl2 (three times with 30 mL each time). The combined organic layers were dried over Na2SO4 and filtered, and the solvent was removed under reduced pressure. No further purification of the product was required. Adapted from reference 13.
Derivative (12), 3,4-dimethoxybenzenesulfonohydrazide was obtained as a white solid with 91% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 8.20 (s, 1H), 7.39 (dd, J =8 Hz, J =2 Hz, 1H), 7.29 (s, 1H, J =2 Hz), 7.11 (d, J =8 Hz, 1H), 4.2 (s, 2H), 3.77 (s, 3H), 3.74 (s, 3H).
Procedure to prepare 3,4-dimethoxy-N-methylbenzenesulfonohydrazide (13)
In a reaction flask, 3,4-dimethoxybenzenesulfonohydrazide (12, 4.31 mmol) and phthalic anhydride (6.45 mmol) were added. The mixture was heated to 130°C without solvent for 2 hours to complete the reaction. The crude solid formed was washed with water with 10% sodium carbonate. Then, the crude solid (3.87 mmol) was added, without a previous purification step, to a stirred acetone suspension with sodium carbonate (4.31 mmol) and ICH3 (8.62 mmol). The mixture was heated to 50°C until the end of the reaction after 18 hours. Next, 50 mL of an aqueous solution of 10% sodium carbonate was added, the layers were separated, and the aqueous phase was extracted with ethyl acetate (three times with 50 mL each time). The organic phase was reduced under negative pressure, obtaining a white solid, which was added to a solution of hydrazine hydrate (17.24 mmol) in ethanol (50 mL) for 5 hours at 80°C until completion of the deprotection step. Then the solvent was reduced under negative pressure, and 30 mL of water with 10% sodium carbonate and CH2Cl2 were added. The layers were separated, and the aqueous phase was extracted (three times with 50 mL of CH2Cl2 each time), and an orange solid with 39% yield was obtained after the concentration step.Citation13
1H NMR (200 MHz, CDCl3) δ (ppm): 7.12 (dd, 1H, J =8 Hz, J =2 Hz), 7.10 (s, J =2 Hz), 6.85 (d, J =8 Hz, 1H), 4.40 (s, 2H), 3.72 (s, 3H), 3.70 (s, 3H), 3.42 (s, 3H).
General procedure for the synthesis of sulfonylhydrazones 4–10
A mixture of the required 3,4-dimethoxybenzenesulfonohydrazide (12) or 3,4-dimethoxy-N-methylbenzenesulfonohydrazide (13) (0.5 mmol) and the appropriate aldehyde (0.5 mmol) was dissolved in ethyl alcohol (20 mL) and was vigorously stirred for 30–120 minutes at room temperature. Once the reaction was completed, the product was precipitated by the dropwise addition of cold water. Following filtration, the precipitate was washed with H2O (10 mL) and n-hexane (10 mL) and dried in vacuum.Citation13
(E)-3-((2-(3,4-dimethoxyphenylsulfonyl) hydrazono)methyl)benzoic acid (4, LASSBio-1775)
Compound 4 (LASSBio-1775) was obtained as a white solid through condensation of 3,4-dimethoxybenzenesulfonohydrazide (12) with methyl 3-formylbenzoic acid with 99% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 11.44 (s, 1H), 8.13 (s, 1H), 7.95 (s, 1H), 7.91 (d, J =7.7 Hz, 1H), 7.76 (d, J =7.7 Hz, 1H), 7.49 (t, J =8.2 Hz, 1H), 7.43 (dd, J =8.2 Hz, 1H), 7.33 (d, J =1.8 Hz, 1H), 7.11 (d, J =8.2 Hz, 1H), 3.78 (s, 6H).
13C NMR (50 MHz, DMSO-d6) δ (ppm): 166.94, 152.52, 148.62, 146.11, 134.24, 131.43, 131.29, 130.67, 130.46, 129.34, 127.04, 121.00, 111,35, 109.95, 55.92, 55.80.
HPLC 60/40 acetonitrile/water: 98.09% purity (Retention Time [RT] =3.15, 254 nm).
(E)-methyl 3-((2-(3,4-dimethoxyphenylsulfonyl)-2-methylhydrazono)methyl)benzoate (5, LASSBio-1853)
Compound 5 (LASSBio-1853) was obtained as a white solid through condensation of 3,4-dimethoxy-N-methylbenzenesul-fonohydrazide (13) with methyl 3-formylbenzoate 62% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 8.26 (s, 1H), 8.00–7.87 (m, 3H), 7.58 (t, J =8 Hz, 1H), 7.44 (dd, J =8, 2.0 Hz, 1H), 7.31 (d, J =2.0 Hz, 1H), 7.14 (d, J =8 Hz, 1H), 3.88 (s, 3H), 3.79 (s, 3H), 3.19 (s, 3H).
13C NMR (50 MHz, DMSO-d6) δ (ppm): 165.77, 152.85, 148.51, 142.82, 134.85, 131.39, 130.16, 129.35, 127.09, 126.96, 121.49, 111.25, 110.21, 55.82, 55.61, 52.27, 33.54.
HPLC 60/40 acetonitrile/water: 97.73% purity (RT =8.12, 254 nm).
(E)-methyl 4-((2-(3,4-dimethoxyphenylsulfonyl)-2-methylhydrazono)methyl)benzoate (6, LASSBio-1774)
Compound 6 (LASSBio-1774) was obtained as a white solid through condensation of 3,4-dimethoxy-N-methylbenzenesulfonohydrazide (13) with methyl 4-formylbenzoate with 75% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 7.99 (d, J =8.2 Hz, 2H), 7.84 (s, 1H), 7.78 (d, J =8.2 Hz, 2H), 7.43 (dd, J =8.4, 2 Hz, 1H), 7.27 (s, J =2 Hz, 1H), 7.12 (d, J =8.4 Hz, 1H), 3.84 (s, 3H), 3.79 (s, 3H), 3.76 (s, 3H), 3.18 (s, 3H).
13C NMR (50 MHz, DMSO-d6) δ (ppm): 165.68, 152.41, 148.50, 145.37, 137.98, 130.37, 130.29, 129.53, 126.78, 120.96, 111.22, 109.73, 55.75, 55.72, 52.15.
HPLC 60/40 acetonitrile/water: 96.68% purity (RT =7.89, 254 nm).
(E)-methyl 4-((2-(3,4-dimethoxyphenylsulfonyl)hydrazono) methyl)benzoate (7, LASSBio-1773)
Compound 7 (LASSBio-1773) was obtained as a white solid through condensation of 3,4-dimethoxybenzenesulfonohydrazide (12) with methyl 4-formylbenzoate with 99% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 11.61 (s, 1H), 7.96 (m, 3H), 7.71 (d, J =8 Hz, 2H), 7.48 (dd, J =8.5, 2.1 Hz, 1H), 7.35 (d, J =2.1 Hz, 1H), 7.14 (d, J =8.5 Hz, 1H), 3.84 (s, 3H), 3.81 (s, 6H).
13C NMR (50 MHz, DMSO-d6) δ (ppm): 165.68, 152.41, 148.50, 145.37, 137.98, 130.37, 130.29, 129.53, 126.78, 120.96, 111.22, 109.73, 55.75, 55.72, 52.15.
HPLC 60/40 acetonitrile/water: 95.47% purity (RT =7.07, 254 nm).
(E)-N′-(4-cyanobenzylidene)-3,4-dimethoxybenzenesulfonohydrazide (8, LASSBio-1771)
Compound 8 (LASSBio-1771) was obtained as a yellow solid through condensation of 3,4-dimethoxybenzenesulfonohydrazide (12) with 4-formylbenzonitrile with 77% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 11.38 (s, 1H), 7.95 (s, 1H), 7.84 (d, J =8.3 Hz, 2H), 7.77 (d, J =8.3 Hz, 2H), 7.46 (dd, J =8.5, 2.0 Hz, 1H), 7.32 (d, J =2.0 Hz, 1H), 7.13 (d, J =8.5 Hz, 1H), 3.80 (s, 3H), 3.79 (s, 3H).
13C NMR (50 MHz, DMSO-d6): δ (ppm) 152.49, 148.54, 144.72, 138.04, 132.68, 130.23, 127.22, 121.06, 118.52, 111.52, 111.89, 111.27, 109.72, 55.80, 55.77.
HPLC 60/40 acetonitrile/water: 99.52% purity (RT =5.00, 254 nm).
(E)-N′-(4-cyanobenzylidene)-3,4-dimethoxy-N-methylbenzene-sulfonohydrazide (9, LASSBio-1772)
Compound 9 (LASSBio-1772) was obtained as a yellow solid through condensation of 3,4-dimethoxy-N-methylbenzenesulfonohydrazide (13) with 4-formylbenzonitrile with 91% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 7.89 (d, J =8.0 Hz, 2H), 7.86 (s, 1H), 7.83 (d, J =8.0 Hz, 2H), 7.45 (d, J =8.5 Hz, 1H), 7.28 (s, 1H), 7.15 (d, J =8.5 Hz, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 3.21 (s, 3H).
13C NMR (50 MHz, DMSO-d6) δ (ppm): 152.96, 148.59, 141.48, 138.64, 132.76, 127.32, 127.05, 121.65, 118.61, 111.69, 111.34, 110.12, 55.85, 55.81, 33.46.
HPLC 60/40 acetonitrile/water: 97.71% purity (RT =7.52, 254 nm).
(E)-4-((2-(3,4-dimethoxyphenylsulfonyl) hydrazono)methyl)benzoic acid LASSBio-1934 (10)
Compound 10 (LASSBio-1934) was obtained as a white solid through condensation of 3,4-dimethoxybenzenesulfonohydrazide (12) with methyl 4-formylbenzoic acid with 99% yield.
1H NMR (200 MHz, DMSO-d6) δ (ppm): 11.53 (s, 1H), 7.93 (s, 1H), 7.91 (d, J =8.4 Hz, 2H), 7.65 (d, J =8.4 Hz, 2H), 7.44 (dd, J =8.6, 2.1 Hz, 1H), 7.32 (d, J =2.1 Hz, 1H), 7.11 (s, J =8.6 Hz, 1H), 3.77 (s, 3H), 3.78 (s, 3H).
13C NMR (50 MHz, DMSO-d6) δ (ppm): 166.95, 152.56, 148.65, 145.76, 137.76, 131.80, 130.43, 129.86, 126.84, 121.16, 111.37, 109.86, 56.15, 55.91.
HPLC 60/40 acetonitrile/water: 98.90% purity (RT =3.05, 254 nm).
Solubility assay
The solubility assay was performed considering the absorptivity of compounds under ultraviolet spectroscopy as described by Schneider et al.Citation18 Assay wavelength was determined through λmax characteristic of each compound. Saturated aqueous solutions were prepared (0.8–1.0 mg/mL) and kept under stirring for 4 hours at 37°C. The supernatant was filtered in 0.45 mm filters and transferred to a quartz cuvette (10 mm) for spectra acquisition. Solubility was determined by linear regression used as Excel graph plots, solutions were prepared by dilutions of the original solution in methanol. Data were collected in triplicate and the mean values were used to create the graph plots. The correlation coefficient (R2) values were between 0.9982 and 1.
Rat plasma stability studies
For conducting these studies, rat plasma was obtained from blood by centrifugation and diluted in phosphate buffer (pH 7.4) to obtain a final plasma concentration of 80%. After that, test compounds were added at a final concentration of 20 µM with 250 µL of final volume and incubated at 37°C for 60 minutes under agitation. At the end of the incubation time, the reaction was stopped by the addition of 450 µL of MeOH and 450 µL of CH3CN. The three independent experiments were performed in duplicate. The samples were centrifuged and filtered for HPLC-UV analysis.Citation20
HPLC-UV analysis
The organic fraction was analyzed with the Shimadzu Prominence HPLC system (Shimadzu, Tokyo, Japan) consisting of a vacuum degasser (DGU-20A5), a binary pump (LC-20AD), an autosampler (SIL-20A), UV/VIS Photodiode Array Detector (SPD-M20A), and fitted with a guard column (CLC G-ODS) and Kromasil 100-5C18 (4.6×250 mm) analytical column running at room temperature. Gradient elution was performed with acetonitrile–water (50:50 v/v – 90:10 v/v – 50:50 v/v), at a flow rate set at 1.0 mL/min. The mobile phase pH was adjusted to 7.0 with NH4OH solution. The detection was carried out at 254 nm wavelength.
HPLC/MS analysis
The organic fraction was analyzed with the Shimadzu Prominence HPLC system (Shimadzu) consisting of a vacuum degasser (DGU-20A5), a binary pump (LC-20AD), an autosampler (SIL-20A), UV/VIS Photodiode Array Detector (SPD-M20A), and fitted with a guard column (CLC G-ODS) and a Shimadzu (CLC-ODS, M) column (250×4.6 mm internal diameter) running at 20°C. Gradient elution was performed with acetonitrile–water (50:50 v/v – 90:10 v/v – 50:50 v/v), at a flow rate set at 1.0 mL/min. The mobile phase pH was adjusted to 7.0 with NH4OH solution. The detection was carried out at 254 nm wavelength. Compounds 7 and 8 and their metabolites (M) were detected with electrospray ionization MS (HPLC-ESI-MS) model Esquire 6000-ESI Ion Trap MSn System Bruker Daltonics (LASSBio®-UFRJ) performed with ESI in positive and negative mode. The capillary voltage was 4.0 mV. The collision energy was set at 25 eV using helium as collision gas. Nitrogen nebulizer gas flow was 4.0 L/min, temperature was 250°C, and pressure was 15 psi. The LC-ESI-MS chromatograms were obtained by scanning over m/z 100–1,000 range.
pKa analysis
The pKa assay was performed considering the absorptivity of compounds in ultraviolet spectroscopy, and the buffer solutions (pH of 3.0–12) were prepared as described by Martínez and Dardonville.Citation19 DMSO stock solutions of the compounds were prepared at 10 mM for the more soluble compounds, and 5 mM for the less soluble compounds. Standard solutions were prepared by adding 4 µL of the stock solution in 196 µL of buffer solution in each well of the microplate, getting a final concentration of 0.2 mM and 0.1 mM analyte, respectively. UV spectra were recorded on a SpectraMAx apparatus between 200 and 500 nm at 2 nm resolution. The data were processed using Excel. pKa values were established by nonlinear regression (GraphPad Prism 5 program).
Chemical stability assays
Chemical stability studies were conducted at two different pH values (2 and 7.4).Citation20 The stock solutions of compounds were prepared at 5 mM to 10 mM concentrations and solubilized in DMSO. Standard solutions were prepared by adding 2 µL of the stock solution in 249 µL acid buffer (0.2 M potassium chloride and HCl 0.2 M; pH =2) or basic (dibasic, pH =7.4) in an Eppendorf microtube. The mixture was placed in a water bath at 37°C under vigorous stirring for 0, 30, 60, and 120 minutes. Once the reaction time was over, 249 µL of basic buffer (phosphate buffer, pH =8.4) was added to neutralize the pH of the medium in experiments using acidic buffer. Extraction of the compound was performed by adding 1.0 mL of acetonitrile. The organic phase was separated, filtered, and analyzed by high-performance liquid chromatography photodiode array detection method (acetonitrile/water mobile phase and 50% to 60%).
Authors contributions
All authors contributed toward data analysis, drafting, and revising the manuscript and agree to be accountable for the work.
Acknowledgments
The authors would like to thank CNPq (BR), FAPERJ (BR), and INCT-INOFAR (BR, 573.564/2008-6 and E-26/170.020/2008) for fellowship and financial support. The authors would also like to thank Matheus da Silva Delgobbo and Dr Carlos Alberto Manssour Fraga.
Disclosure
The authors report no conflicts of interest in this work.
References
- VevesABackonjaMMalikRAPainful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment optionsPain Med20089666067418828198
- VinikAINevoretMLCaselliniCParsonHDiabetic neuropathyEndocrinol Metab Clin North Am201342474778724286949
- DothAHHanssonPTJensenMPTaylorRSThe burden of neuropathic pain: a systematic review and meta-analysis of health utilitiesPain2010149233834420227832
- BrodMBlumSIBushnellDMRamasamyADevelopment and validation of the Diabetic Peripheral Neuropathic Pain Impact (DPNPI) measure, a patient-reported outcome measureQual Life Res201524123001301426068732
- BrodMPohlmanBBlumSIRamasamyACarsonRBurden of Illness of Diabetic Peripheral Neuropathic Pain: A Qualitative StudyPatient20158433934825354872
- TesfayeSBoultonAJDyckPJDiabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatmentsDiabetes Care201033102285229320876709
- PeltierAGoutmanSACallaghanBCPainful diabetic neuropathyBMJ2014348g179924803311
- SnedecorSJSudharshanLCappelleriJCSadoskyAMehtaSBottemanMSystematic review and meta-analysis of pharmacological therapies for painful diabetic peripheral neuropathyPain Pract201414216718423534696
- FreemanRNew and developing drugs for the treatment of neuropathic pain in diabetesCurr Diab Rep201313450050823771401
- FreitagCMMillerRJPeroxisome proliferator-activated receptor agonists modulate neuropathic pain: a link to chemokines?Front Cell Neurosci2014823825191225
- AmbrosinoPSoldovieriMGMariaMRussoCTaglialatelaMFunctional and biochemical interaction between PPARα receptors and TRPV1 channels: Potential role in PPARα agonists-mediated analgesiaPharmacol Res20148711312225014183
- Menéndez-GutiérrezMPRoszerTRicoteMBiology and therapeutic applications of peroxisome proliferator-activated receptorsCurr Top Med Chem201212654858422242855
- Zapata-SudoGLimaLMPereiraSLDocking, synthesis and anti-diabetic activity of novel sulfonylhydrazone derivatives designed as PPAR-gamma agonistsCurr Top Med Chem201212192037204823167793
- LimaLMTrachezMMde AraujoJSNovel Partial Agonist of PPAR-Gamma for Treatment of Diabetic Neuropathy in RatsJ Diabetes Metab20145392
- PallaGPredieriGDomianoPVignaliCTurnerWConformational behaviour and E/Z isomerization of N-acyl and N-aroylhydrazonesTetrahedron19864236493654
- RodriguesDAFerreira-SilvaGÀFerreiraACDesign, Synthesis and Pharmacological Evaluation of Novel N-Acylhydrazone Derivatives as Potent Histone Deacetylase 6/8 Dual InhibitorsJ Med Chem201659265567026705137
- da SilvaTFBispo JúniorWAlexandre-MoreiraMSNovel orally active analgesic and anti-inflammatory cyclohexyl-N-acylhydrazone derivativesMolecules20152023067308825685912
- SchneiderPHosseinySSSzczotkaMJordanVShlitterKRapid solubility determination of the triterpenes oleanoic acid and ursolic acid by UV spectroscopy in different solventsPhytochem Lett200928587
- MartínezCHDardonvilleCRapid Determination of Ionization Constants (pK a) by UV Spectroscopy Using 96-Well Microtiter PlatesACS Med Chem Lett20134114214524900577
- AlvesMAde QueirozACAlexandre-MoreiraMSDesign, synthesis and in vitro trypanocidal and leishmanicidal activities of novel semicarbazone derivativesEur J Med Chem2015100243326069927
- KazakevichYLobruttoRHPLC for Pharmaceutical Scientistis1st edNew YorkWiley2007