
Abstract
The identification of echinoderm microtubule-associated protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) fusion gene in non-small cell lung cancer (NSCLC) has radically changed the treatment of a subset of patients harboring this oncogenic driver. Crizotinib was the first ALK tyrosine kinase inhibitor to receive fast approval and is currently indicated as the first-line therapy for advanced, ALK-positive NSCLC patients. However, despite crizotinib’s efficacy, patients almost invariably progress, with the central nervous system being one of the most common sites of relapse. Different mechanisms of acquired resistance have been identified, including secondary ALK mutations, ALK copy number alterations and activation of bypass tracks. Different highly potent and brain-penetrant next-generation ALK inhibitors have been developed and tested in NSCLC patients with ALK rearrangements. Ceritinib, a structurally distinct and selective ALK inhibitor, showed 20 times higher potency than crizotinib in inhibiting ALK and had activity against the most common crizotinib-resistant mutations, including L1196M and G1269A, in preclinical models. In Phase I and II studies, ceritinib demonstrated pronounced activity in both crizotinib-naïve and crizotinib-refractory patients, with responses observed regardless of the presence of ALK resistance mutations. Ceritinib was the first ALK inhibitor to be approved for the treatment of crizotinib-refractory, ALK-rearranged NSCLC, and recent results from a Phase III study have demonstrated superior efficacy compared to standard chemotherapy in the first- and second-line setting. We provide an extensive overview of ceritinib from the design of the compound through preclinical data until efficacy and toxicity results from Phase I–III clinical studies. We review the molecular alterations associated with resistance to ceritinib and highlight the importance of obtaining tumor biopsy at progression to tailor therapy based upon the underlying resistance mechanism. We finally provide an outlook on novel rational therapeutic combinations.
Introduction
Lung cancer represents the leading cause of cancer-related mortality worldwide and is responsible for nearly one cancer death in five among both sexes.Citation1 Non-small cell lung cancer (NSCLC), the most common type of lung cancer, includes adenocarcinoma, squamous cell carcinoma and large-cell carcinoma histosubtypes and is generally diagnosed at advanced stages. Systemic therapy is the standard of care for advanced NSCLC patients and, until recently, platinum-based chemotherapy has been the only available first-line therapeutic option. Bevacizumab, a monoclonal antibody anti-vascular endothelial growth factor, was approved in combination with first-line platinum-based chemotherapy based on modest overall survival (OS) improvement in patients with nonsquamous NSCLC.Citation2,Citation3 Another targeted agent, the anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibody necitumumab, has been approved with gemcitabine and cisplatin as the first-line treatment of metastatic squamous NSCLC based on a 1.6-month survival increase for the combination compared to chemotherapy alone.Citation4 However, there are no current validated predictive biomarkers to select patients for both antiangiogenic drugs and anti-EGFR monoclonal antibodies.
The discovery in some subsets of NSCLC, mainly adenocarcinoma, of a number of actionable oncogenic alterations, including EGFR mutations and ALK rearrangements, conferring unique sensitivity to inhibition by specific molecularly targeted agents, has opened a new era of personalized medicine for lung cancer. Based on recent developments, NSCLC is not anymore considered a single entity, but a heterogeneous disease including morphologically and molecularly distinct subsets of tumors characterized by different therapeutic vulnerabilities.Citation5 The tyrosine kinase inhibitors (TKIs) targeting EGFR and ALK have consistently demonstrated superior efficacy compared to chemotherapy, and they currently represent the standard of care of these molecularly defined subgroups of NSCLC patients.Citation6–Citation8 Therefore, besides histopathologic assessments, molecular profiling of lung cancer has been rapidly incorporated into the diagnostic process to guide treatment decisions.Citation9
EML4/ALK in NSCLC
The identification of the echinoderm microtubule-associated protein-like 4 (EML4)–anaplastic lymphoma kinase (ALK) fusion gene as an oncogenic driver in NSCLC in 2007 revolutionized the therapeutic management and improved the prognosis of the relatively small, but still relevant, subset of patients harboring this molecular alteration.Citation10 The EML4–ALK fusion gene arises from a small inversion within the short arm of chromosome 2 that joins the 5′-end (encoding the NH2-terminal portion, including the coiled-coil domain) of the EML4 gene to the 3′-end (encoding the COOH-terminal portion, including the tyrosine kinase domain) of the ALK gene. The ALK gene encodes for a protein of 1,620 amino acids that is a transmembrane tyrosine kinase receptor belonging to the insulin receptor superfamily. ALK has a probable role in the normal development and function of the nervous system.Citation11–Citation13 In adult human tissues, expression of ALK appears restricted to certain neuronal cells. Pleiotrophin and midkine have been postulated to be the activating ligands of ALK. Both factors have shown to induce neuronal growth, but are also implicated in other processes such as cell migration and angiogenesis.Citation13–Citation15 Gene amplification, activating mutations and chromosomal translocations with the formation of fusion genes may be responsible for ALK activation in tumor cells.Citation16
The EML4–ALK fusion gene results in a chimeric oncoprotein that undergoes constitutive dimerization and activation of the tyrosine kinase function of ALK and its downstream signaling, including Ras/mitogen-activated protein kinase (MAPK), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (AKT) and Janus kinase (JAK)/signal transducer and activator of transcription path-ways, which can promote cell proliferation, differentiation, and provide antiapoptotic signals. The ALK fusion product was demonstrated to be oncogenic in lung cancer and to drive transformation both in vitro and in vivo, thus suggesting its inhibition could represent an effective therapeutic strategy.Citation17,Citation18 Multiple variants of EML4–ALK have been reported, at least 15, depending on different breakpoints in the EML4 fused to the kinase domain of ALK. The most common of them include variant 1, where exon 13 of EML4 is fused to exon 20 of ALK (E13;A20), variant 2, where exon 20 of EML4 is fused to exon 20 of ALK (E20;A20), and variant 3, where exon 6 of EML4 is fused to exon 20 of ALK (E6;A20). Two isoforms for V3 can be generated by alternative splicing, V3a (E6a;A20) and V3b (E6b;A20).Citation19–Citation21 Although the portion of EML4 fused to the kinase domain of ALK can vary, all the variants contain the amino-terminal coiled-coil domain within EML4, which is necessary for the oncogenic activity of EML4/ALK via homodimerization and kinase activation.
Preclinical and clinical data suggest that EML4–ALK variants have different sensitivity to ALK inhibition.Citation22–Citation24 In a retrospective analysis, Yoshida et al showed a better efficacy of crizotinib in terms of progression-free survival (PFS) in patients with ALK variant 1 vs nonvariant 1. Moreover, a greater proportion of patients with ALK variant 1 achieved disease control than those with nonvariant 1.Citation24 The ALK gene can have different fusion partners beyond EML4, including kinesin family member 5b (KFIF5), TRK-fused gene (TGF), kinesin light chain 1 (KLC1), huntingtin-interacting protein 1 (HIP1) and baculoviral inhibition of apoptosis protein repeat containing 6 (BIRC6).Citation24–Citation30
Rearrangements of the ALK gene have been identified in ~3%–7% of NSCLC and are more frequent in younger patients, with adenocarcinoma histology with signet-ring cell pattern and a never or light smoking history.Citation31,Citation32 ALK rearrangements generally occur independently of other driver mutations, including EGFR and KRAS mutations,Citation33 although concomitant actionable mutations have been described. Several methods are currently available to detect ALK rearrangements in NSCLC clinical samples.Citation34–Citation37 The break-apart fluorescence in situ hybridization was the first diagnostic test to be approved by the US Food and Drug Administration (FDA) as a diagnostic gold standard for screening of ALK-rearranged NSCLC. Other methods include immunohistochemistry, demonstrating high sensitivity, specificity and comparable results with fluorescence in situ hybridization, although it is less expensive and time consuming, real-time polymerase chain reaction, and novel techniques such as hybrid capture-based next-generation sequencing and Nanostring technology.
Current treatment of ALK-rearranged NSCLC
Crizotinib, an oral small-molecule TKI of ALK, c-MET and ROS1 kinases,Citation38,Citation39 was the first targeted agent to be approved in 2011 for pretreated, ALK-rearranged NSCLC, based on pronounced activity seen in early Phase I and II studies, with the objective response rates (ORRs) ranging from 57% to 61% and median PFS from 8.1 to 9.7 months.Citation40–Citation42 Subsequent Phase III trials demonstrated the superiority, in terms of PFS and ORR, of crizotinib compared to standard chemotherapy in previously treated (PROFILE 1007 study)Citation43 and previously untreated (PROFILE 1014 study)Citation44 patients with advanced ALK-positive NSCLC, thus establishing it as a standard of care for this subset of patients also in the first-line setting. In both studies, crizotinib was generally well tolerated and resulted in greater improvement in quality of life (QoL) compared to chemotherapy.Citation43,Citation44
However, despite marked and durable initial responses to crizotinib, most patients develop progressive disease, generally within 1–2 years of starting therapy, similar to what happens with other targeted therapies.Citation45 The central nervous system (CNS) is one of the most common sites of relapse. This can be explained by pharmacokinetic (PK) limitations rather than a biologic resistance. Crizotinib is indeed a known substrate of P-glycoprotein (P-gp), a drug efflux pump expressed in the capillary epithelial cells of the blood–brain barrier (BBB) that limits accumulation of the drug in the CNS.Citation46–Citation48 Molecular mechanisms leading to crizotinib resistance have been commonly classified as on-target genetic alterations (secondary point mutations within the ALK-TK domain and ALK copy number alterations) and off-target mechanisms, such as activation of “bypass” signaling pathways and others (eg, epithelial–mesenchymal transition).Citation49–Citation51
The initial report of two secondary mutations (L1196M and C1156Y) came from the molecular analysis of postprogression tissue biopsy from a patient who developed resistance to crizotinib after 5 months.Citation52 The gatekeeper mutation L1196M interferes with crizotinib binding through steric hindrance and is analogous to T315I in the BCR–ABL fusion gene and T790M in the EGFR gene conferring resistance to corresponding TKIs. The G1269A mutation is also located in the adenosine triphosphate (ATP)-binding pocket and affects ALK TKI binding. Other secondary resistance mutations have been identified from molecular characterization of postprogression patient specimens (occurring in ~20%–40% of cases) or ALK-resistant cell lines (derived from patients or generated in vitro), including C1156Y, F1174, 1151Tins, L1152R, S1206Y, I1171T, G1202R and D1203N.Citation49–Citation51,Citation53–Citation55 Multiple nonoverlapping mutations within the ALK TK domain have been reported, and also, ALK copy number alterations (copy number gain or amplification) can coexist in some cases, thus suggesting a potential heterogeneity of biologic resistance.Citation50,Citation51,Citation56 Activation of alternate kinases, including EGFR, KIT, human epidermal growth factor receptor 2 (HER2)/HER3, insulin-like growth factor 1 receptor (IGF-1R), Rous sarcoma oncogene cellular homologue (SRC) and MAPK, leads to ALK-independent growth and resistance to inhibition by crizotinib.Citation49,Citation57–Citation61
The evidence of a lack of long-term benefit from crizotinib due to acquisition of resistance has prompted the development of increasingly potent, selective and brain-penetrant ALK inhibitors, with differential spectrum of activity against the most common resistance mutations. Indeed, in initial studies in preclinical models, Ba/F3 cells expressing wild-type EML4–ALK or EML4–ALK with resistance mutations were tested for their sensitivity to a panel of ALK inhibitors.Citation51 All of the mutant forms of EML4–ALK were found to be resistant to crizotinib compared with wild-type EML4–ALK and showed different degrees of sensitivity to next-generation ALK inhibitors depending on the type of mutation. For example, G1269A substitution was sensitive to several second-generation ALK inhibitors, whereas G1202R, at the solvent front of the kinase domain of ALK, conferred high-level resistance to almost all the ALK TKIs tested.
Different next-generation ALK inhibitors are currently being evaluated in clinical studies and some have demonstrated pronounced systemic and intracranial activity, in both crizotinib-resistant and crizotinib-naïve ALK-rearranged NSCLC. Ceritinib was the first next-generation ALK inhibitor to be approved by the FDA in 2014 for the treatment of patients with ALK-positive metastatic NSCLC who have progressed on or are intolerant to crizotinib and then by the European Medicines Agency (EMA) in 2015 for advanced ALK-positive NSCLC patients previously treated with crizotinib. Among the other next-generation ALK inhibitors, alectinib is currently approved by the FDA and EMA for the treatment of crizotinib-pretreated patients and brigatinib received “Breakthrough Therapy” designation from the FDA for the same indication.
Ceritinib: overcoming crizotinib resistance
Design and mechanism of action
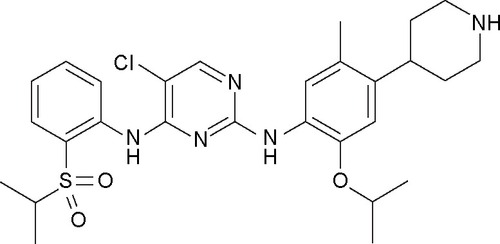
Ceritinib (LDK378; Zykadia®, Novartis Pharmaceuticals Corporation) is a potent and selective oral, ATP-competitive TKI of ALK. Ceritinib was derived from the optimization of a first lead compound identified by Novartis, TAE684,Citation62 which was shown to be a potent and specific ALK inhibitor, but to have some deficiencies such as the capability to form an extensive number of reactive adducts upon metabolic oxidation, which might create the potential for toxicologic liabilities. This was mainly correlated with the presence of a solubilizing group connected by a nitrogen atom into the central aniline moiety. The modification of this molecule to design novel derivatives was thus performed by reversing the piperidine at the para position of the aniline moiety, replacing the methoxy moiety by an isopropoxy moiety and including a methyl group at the position para to the isopropoxy moiety.Citation63,Citation64 Through these modifications, a potent and selective ALK inhibitor, 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl) phenyl)pyrimidine-2,4-diamine (LDK378, ceritinib), was synthesized (). The molecular formula of ceritinib is C28H36ClN5O3S and the molecular weight is 558.14 Da. In an enzymatic kinase assay, ceritinib showed higher potency than crizotinib against ALK (IC50 value of 200 pM) and, among a panel of 30 kinases, it also showed biochemical inhibition of IGF-1R, InsR and STK22D. When the cellular kinase pro-file of ceritinib was assessed in Ba/F3 cells, it showed high selectivity against ALK (IC50 of 2.2 nM), without inhibiting the activity of other kinases, including IGF-1R. However, inhibition of IGF-1R and other kinases, including InsR and ROS1, can occur at clinically relevant concentrations.Citation65,Citation66 In Ba/F3 cells transfected with the NPM–ALK fusion gene, ceritinib showed potent antiproliferative activity (IC50 value of 26 nM), with good selectivity over wild-type Ba/F3 cells (IC50 >2 μM) and Ba/F3 cells transfected with Tel-InsR gene (IC50 =320 nM).Citation63 Ceritinib had an excellent PK profile in rodents and nonrodents, with an oral bioavailability of >50%. In in vivo studies, the compound displayed significant antitumor activity against rat xenograft models of NPM–ALK anaplastic large cell lymphoma and EML4/ALK-positive NSCLC, causing dose-dependent inhibition of tumor growth, with good tolerability at all dose levels tested.Citation63 In a tissue distribution study using a rat model, ceritinib crossed the BBB with a brain-to-blood exposure (AUCinf) ratio of ~15%.Citation66
Figure 1 Chemical structure of ceritinib.
Preclinical activity of ceritinib against ALK resistance mutations
The activity of ceritinib in preclinical models of ALK-positive lung cancer with acquired resistance to crizotinib was examined in a landmark study by Friboulet et al.Citation67 In in vitro enzymatic assays, ceritinib was 20-fold more potent against ALK than crizotinib. Moreover, it was more potent than crizotinib against ALK-positive cancer cell lines (H3122 and H228), which led to suppression of ALK phosphorylation, as well as suppression of downstream pathways (such as PI3K/AKT, MAPK/Erk kinase/extracellular signal-regulated kinase and mammalian target of rapamycin) at lower doses than crizotinib (). In in vivo studies in treatment-naïve H2228 xenograft models, ceritinib showed marked and more durable antitumor activity than crizotinib. Ceritinib demonstrated activity against crizotinib-resistant mutations in different cancer cell lines, including two cell lines harboring the L1196M and G1269M mutations that were established from biopsies of ALK-positive NSCLC patients who had become resistant to crizotinib, with GI50 (50% growth inhibition) values from 6- to 36-fold inferior compared to crizotinib.Citation67 In crizotinib-resistant xenograft models derived from cells harboring the L1196M mutation, ceritinib was able to control tumor growth at lower doses compared to crizotinib. Notably, a cell line without any detectable ALK alterations, derived from a biopsy of a patient at the time of progression to crizotinib was highly sensitive to ceritinib, supporting the clinical observation that ceritinib was highly active in crizotinib-resistant cancers with or without ALK secondary mutations.Citation67
Figure 2 Ceritinib is a potent and selective ALK tyrosine kinase inhibitor, with activity against various crizotinib and alectinib resistance mutations (in green). Ceritinib-resistant ALK mutations (in red) include G1202R and F1174C/L.
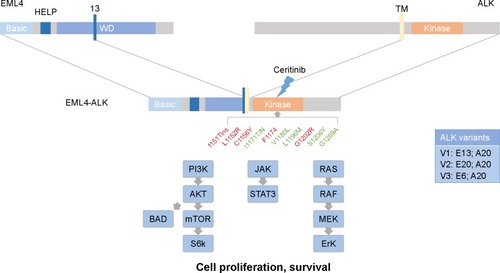
The activity of ceritinib against nine additional ALK mutations conferring resistance to crizotinib was tested in Ba/F3 cells. Ceritinib potently inhibited the growth of those cells expressing the most common mutations, L1196M, G1269A and also S1206Y and I1171T, but not those with C1156Y, G1202R, 1151Tins, L1152R and F1174C mutations, although it was still more potent than crizotinib against these less-common mutations (). In in vivo studies, ceritinib suppressed tumor growth in multiple crizotinib-resistant xenograft tumor models, with impressive antitumor activity in I1171T-resistant models, while less and no activity was observed in the C1156Y- and G1202R-resistant models, respectively.Citation67
One main reason explaining the ability of ceritinib to overcome most crizotinib-resistant mutations relies on the distinct molecular structure of these two compounds. Indeed, the presence of G1269A does not prevent ceritinib binding, while, on the other hand, it creates a steric hindrance to the phenyl ring of crizotinib. In the presence of the gatekeeper mutation L1196M, the Cl-moiety of the pyrimidine hinge-binding core of ceritinib interacts with methionine (Met). In contrast, crizotinib binding is affected through both steric interference and unfavorable interactions with the 2-amino substituent of the pyridinyl hinge-binding core and the methyl substituent of the alkoxy moiety of crizotinib.Citation67 Conversely, ceritinib was less active against the other less-common mutations, including G1202R. The G1202R substitution is located at the solvent front of the ALK domain, abutting the crizotinib-binding site and likely diminishing the binding affinity of all other ALK inhibitors, including ceritinib, to the mutant kinase for steric hindrance, due to the presence of a large, bulky basic residue.Citation51
In summary, the next-generation ceritinib was able to overcome various crizotinib-resistant mutations and was potent against several in vitro and in vivo laboratory models of acquired resistance to crizotinib.
Pharmacokinetics
The PK profile of ceritinib was first evaluated in mice, rats, dogs and monkeys and found to be consistent across all these species. The compound showed good oral bioavailability (≥55%) following a single oral administration and a low plasma clearance compared to liver blood flow, with a high volume of distribution at a steady state (Vss). The time to maximum plasma concentration (Tmax) occurred late after oral administration, indicating slow oral absorption, with a half-life (T1/2) ranging from moderate to long (6.2–26 hours).Citation63
PK data in humans were reported from patients treated with ceritinib in the first-in-human study. The ASCEND-1 trial, enrolling patients with advanced cancer harboring genetic alterations in ALK (the majority were advanced NSCLC), included a dose-escalation phase, determining the maximum tolerated dose (MTD), followed by a dose-expansion phase.Citation68 PK analyses were based on data from the dose-escalation phase, in which patients were treated with single daily oral ceritinib at dose levels of 50–750 mg daily, continued in 21-day cycles. Results from these analyses showed that exposure to ceritinib increased with the dose. The maximal plasma concentration (Cmax) increased slightly more than proportional to the dose, across daily doses ranging from 50 to 750 mg. During the 3-day PK evaluation period, after the administration of the first dose of ceritinib at the established MTD (750 mg daily), the Cmax of ceritinib was achieved ~6 hours later and the mean terminal half-life was ~40 hours. The mean (±SD) area under the plasma concentration–time curve (AUC) over a 24-hour period on day 8 was 16,500±4,750 ng/mL/hour. The mean Cmax was 800±205 ng/mL. After multiple repeated daily oral doses, steady-state levels of ceritinib were achieved by ~day 15.Citation68 The absolute bioavailability of oral ceritinib is not known.Citation66 Two studies were conducted in healthy adults to investigate the influence of food on the oral bioavailability of ceritinib: a study with low- or high-fat meals at 500 mg and another study with a light snack at 750 mg.Citation69 Higher plasma concentrations for ceritinib were achieved when it was administered under fed conditions (low- and high-fat meals). The Cmax and the AUC from time zero to infinity (AUC0–∞) of ceritinib were increased by 43% and 58% after a low-fat meal and by 41% and 73% after intake of a high-fat meal, respectively, compared to fasting conditions.Citation69 Similar results were observed with oral administration of 750 mg ceritinib with a light snack. These data suggest a higher oral bioavailability and drug exposure with food, and that ceritinib should be taken on an empty stomach. However, a three-arm randomized trial is ongoing to evaluate an alternative way to give ceritinib (lower doses, 450 or 600 mg taken with a low-fat meal), compared to ceritinib 750 mg taken in a fasted state, that may lead to better gastrointestinal tolerability in patients with ALK-positive NSCLC while maintaining similar steady-state exposure (NCT02299505). Ceritinib is highly bound to plasma protein (about 97%), independent of the drug concentration, and displays a preference for red blood cells over plasma (mean blood-to-plasma ratio of 1.35).Citation66 The volume of distribution (Vd/F) after single oral dose (750 mg) of ceritinib is 4230 L. Ceritinib is primarily metabolized by cytochrome P450 family 3 subfamily A (CYP3A) enzymes and is primarily excreted via feces (92.3%, with 68% of the dose excreted unchanged, vs 1.3% via urine).
Compared with crizotinib, according to the FDA prescribing information,Citation66 the half-life of ceritinib after a single dose is similar (for ceritinib at 750 mg: 41 hours; for crizotinib at 250 mg: 42 hours) and both ALK inhibitors have a lower clearance at steady state after daily dose administration (ceritinib: 33.2 L/hour; crizotinib: 60 L/hour) compared to a single dose (ceritinib: 88.5 L/hour; crizotinib: 100 L/hour), indicating nonlinear PK over time.
Drug interactions
Ceritinib is a substrate of CYP3A, as shown by in vitro studies.Citation66 Coadministration with ketoconazole (a strong CYP3A inhibitor) increased ceritinib exposure by 2.9-fold, and coadministration with rifampin (a strong CYP3A inducer) decreased ceritinib exposure by 70% in healthy subjects.Citation66,Citation70 Ceritinib may inhibit CYP3A and cytochrome P450 family 2 subfamily C member 9 (CYP2C9) at clinical concentrations. Ceritinib is not a substrate of Breast Cancer Resistance Protein (BCRP), Organic Cation Transporter (OCT1), Organic Anion Transporter (OAT2) or Organic Anion Transporter Polypeptide (OATP1B1) transporter proteins, but is a substrate of efflux transporter P-gp; thus, coadministration with drugs inhibiting P-gp may result in increased levels of ceritinib.Citation66 Notably, recent data indi-cate an important role of the multidrug transporters p-gp/ATP-binding cassette subfamily B member 1A (ABCB1) and BCRP/ATP-binding cassette subfamily G member 2 (ABCG2) in regulating accumulation of ceritinib in the brain, thus suggesting a potential combined approach with inhibitors of these proteins and ceritinib to improve treatment of brain metastases or in those tumors overexpressing ABCB1 and/or ABCG2.Citation71 Ceritinib demonstrates poor solubility as pH increases in vitro, thus its bioavailability may be altered by concomitant gastric acid reducing agent administration, although there are no studies specifically designed to evaluate this association.Citation66
Clinical development
Phase I and II studies
Ceritinib was tested in a Phase I study, the ASCEND-1, which enrolled patients with advanced cancers with genetic alterations in ALK, including ALK-rearranged NSCLC (including patients who had previously received an ALK inhibitor).Citation68 In the dose-escalation phase of the study, ceritinib was administered as a single oral daily dose starting from 50 mg, on the basis of preclinical safety data. Based on the occurrence of dose-limiting toxicities, including diarrhea, vomiting, dehydration, elevated aminotransferase levels and hypophosphatemia, the MTD of ceritinib was defined to be 750 mg once daily, which was the dose assessed in the dose-expansion phase. The assessment of antitumor activity of ceritinib was a key secondary objective of this study. Ceritinib demonstrated significant clinical activity in advanced, ALK-rearranged NSCLC patients, with an ORR of 58% (95% CI: 48–67) among those patients who received at least 400 mg of ceritinib per day ().Citation68 The ORR was 62% in ALK inhibitor-naive patients and 56% in crizotinib-pretreated patients, including patients with untreated brain CNS lesions after crizotinib progression. Some responses were dramatic and rapid. The median duration of response was 8.2 months. Overall median PFS was 7 months (95% CI: 5.6–9.5), 6.9 months in the subgroup of ALK inhibitor-pretreated patients and 10.4 months in patients who had not received crizotinib previously. Adverse events (AEs) were more gastrointestinal, of grade 1 or 2 (). Of interest, responses to ceritinib were observed regardless of the presence of ALK resistance mutations detected in tumor tissues from rebiopsy taken at the time of crizotinib resistance before study entry, thus confirming results from preclinical studies and suggesting that cells can become resistant due to incomplete inhibition of ALK by crizotinib at clinically relevant concentrations, so a more potent and structurally distinct ALK inhibitor can overcome this resistance, or even that ceritinib may have a broader range of activity against unknown resistance genetic alterations. Based on this study, in April 2014, the FDA granted accelerated approval to ceritinib for the treatment of patients with ALK-positive metastatic NSCLC with disease progression or who are intolerant to crizotinib. In February 2015, the EMA Committee for Medicinal Products for Human Use adopted a positive opinion, recommending the granting of a conditional marketing authorization for ceritinib in patients with advanced ALK-positive NSCLC previously treated with crizotinib. An updated analysis of the ASCEND-1, including all ALK-rearranged NSCLC patients (n=246) who received oral ceritinib at 750 mg/day in the dose-escalation and -expansion phases, has been recently reported.Citation72 After a median follow-up of 11 months, ceritinib confirmed its high activity and resulted in clinically meaningful and durable antitumor responses in ALK inhibitor-naïve patients (median duration of response [DOR] was 17.0 months and PFS was 18.4 months) and in ALK inhibitor-pretreated patients (median DOR was 8.3 months and PFS was 6.9 months).Citation72 All those patients with tumors harboring common ALK resistance mutations, including E1129V, L1196M, C1156Y, I1171T and F1174V, had partial responses with ceritinib, while a patient with G1202R did not respond.Citation73 Ceritinib also showed significant intracranial responses, as demonstrated in a retrospective analysis. Indeed, of 36 patients with asymptomatic measurable brain metastases at baseline, 63% (95% CI: 25–92) of crizotinib-naïve patients achieved an intracranial response, as did 36% (19–56) of crizotinib-pretreated patients ().Citation73 The median time to intracranial response was similar to that of whole-body response. Responses were observed in more than half of patients who had received prior brain radiotherapy, indicating a BBB penetration of this highly potent ALK inhibitor.
Table 1 Summary of data from selected clinical trials of ceritinib
Table 2 Most common adverse events from clinical trials of ceritinib
In the open-label, multicenter, Phase II study, ASCEND-2, patients with advanced ALK-rearranged NSCLC, previously treated with at least one platinum-based chemotherapy and who experienced progression during crizotinib treatment as their last prior therapy, received oral ceritinib at a standard dose of 750 mg daily.Citation74 Patients with asymptomatic or neurologically stable baseline brain metastases were included. The results from this study were generally consistent with those reported for the ASCEND-1 study. The ORR was 38.6%, with a disease control rate (DCR) of 77.1%. The responses were rapid (time to response [TTR] 1.8 months) and durable (median DOR 9.7 months). The median PFS and OS were 5.7 and 14.9 months, respectively (). Among 20 patients who had baseline measurable brain lesions, an intracranial DCR of 80% was achieved. Overall, the safety profile was consistent with that of ASCEND-1 and no new or unexpected serious AEs (SAEs) were reported ().Citation74 A trend toward improved lung cancer symptoms was observed. However, patients reported worse gastrointestinal symptoms, compared to baseline, throughout the treatment, but the global QoL score was maintained during treatment.
In the ASCEND-3, a single-arm, open-label, multi-center Phase II study, ceritinib was assessed in advanced or metastatic ALK+ NSCLC patients, who were chemotherapy naïve or had received up to three lines of chemotherapy and no prior ALK-TKIs treatment. Long-term follow-up results (>2 years) have been recently presented.Citation75 A total of 124 patients were enrolled; ~40% had brain metastases at baseline, of whom more than a half had received prior brain radiotherapy. The ORR assessed by blinded independent review committee was 63.7% (95% CI: 54.6%–72.2%), with a median PFS of 18.4 months (). Median OS was not reached at data cutoff, and the estimated 24-month OS rate was 67.5%. Intracranial response rate in those patients with measurable brain metastases at baseline (13 patients) was 61.5%, including one complete response, with an intracranial DCR of 76.9%. The AEs were consistent with the known safety profile of ceritinib (). Approximately 80% of patients had dose interruptions or adjustments due to AEs. Updated patient-reported outcome (PRO) results were consistent with the primary analysis and showed improvement of symptoms from baseline and the QoL was maintained.
Phase III studies
Two randomized Phase III trials compared ceritinib vs standard chemotherapy in the first-line (ASCEND-4)Citation76 or second-line (ASCEND-5) setting.Citation74 The primary endpoint of both studies was PFS. The ASCEND-4 trial included 376 patients with previously untreated, ALK-positive, advanced NSCLC, who were randomly assigned to receive ceritinib 750 mg/day or pemetrexed-platinum chemotherapy, followed by maintenance pemetrexed.Citation76 Ceritinib treatment significantly improved median PFS compared to chemotherapy, with a risk reduction of 45% in PFS (16.6 vs 8.1 months for ceritinib and chemotherapy, respectively; hazard ratio [HR] 0.55, 95% CI: 0.42–0.73; P<0.00001), as shown in . Ceritinib was also associated with improved median PFS compared to chemotherapy both in the subgroup of patients without brain metastases (26.3 vs 8.3 months, HR 0.48, 95% CI: 0.33–0.69) and with brain metastases (10.7 vs 6.7 months, HR 0.70, 95% CI: 0.44–1.12). In addition, significantly higher and durable responses were attained with ceritinib compared to chemotherapy (ORR and DOR: 72.7% and 23.9 months vs 27.3% and 16.6 months, respectively), as shown in . In patients with measurable brain metastases at baseline, ceritinib showed a high overall intracranial response rate of 72.7%, an intracranial clinical benefit rate of 86.4% at 24 weeks and a median duration of intracranial response of 16.6 months. The median OS was not reached in the ceritinib group and was 26.2 months in the control group. AEs on ceritinib were frequent, but generally considered manageable and acceptable, and only 5.3% patients discontinued treatment due to AEs suspected to be drug related (). Ceritinib led to significant improvements in QoL and improved lung cancer symptom scores, compared to chemotherapy. These data strongly favor ceritinib vs standard chemotherapy as first-therapeutic approach in ALK-rearranged NSCLC patients. However, given the superiority of crizotinib compared with platinum/pemetrexed chemotherapy in the first-line setting of ALK-positive NSCLC, currently, chemotherapy should not represent the optimal control arm anymore, and current trials are ongoing to compare head-to-head next-generation ALK inhibitors and crizotinib.
In the open-label, randomized, Phase III ASCEND-5 study, ceritinib was compared with chemotherapy (docetaxel or pemetrexed) in locally advanced or metastatic, ALK-rearranged NSCLC patients who had received previous crizotinib (any time prior to enrollment) and chemotherapy (including a platinum doublet).Citation77 Patients who discontinued chemotherapy due to disease progression could crossover to ceritinib. Results were recently presented and showed that median PFS, as assessed by blinded independent review committee, was significantly improved with ceritinib compared to chemotherapy (5.4 vs 1.6 months; HR 0.49, 95% CI: 0.36–0.67; P<0.001). The improvement in PFS was robust, demonstrating consistency across a number of subgroups, and clinical benefit was further supported by ORR (39.1% vs 6.9%) and DCR (76.5% vs 36.2%), as shown in . Furthermore, there was a trend toward a reduction in many patient-reported lung cancer-related symptoms with ceritinib.Citation77 Although the OS data were immature, there seemed to be no improvement with ceritinib, probably because of the high number of patients crossed over to ceritinib after progression. The incidence and the type of AEs were consistent with those reported in Phase I and Phase II study ().
Intracranial activity of next-generation ALK inhibitors in crizotinib-resistant NSCLC patients
A significant portion, reported as 26% of patients enrolled on PROFILE 1014, of patients with ALK-positive NSCLC present with brain metastasis at baseline.Citation44,Citation49 Moreover, CNS is recognized to be one of the most common sites of progression during crizotinib. In a retrospective, pooled analysis from the PROFILE 1005 and 1007 trials, among patients with asymptomatic untreated brain metastases who received crizotinib, the median time to intracranial progression was 7 months, compared with a 12.5-month median time to systemic progression. Moreover, the CNS was a site of progression in 70% of patients with known brain metastases during crizotinib treatment and 20% of those without brain metastases at study entry had progression in CNS on crizotinib.Citation49,Citation78 As we have commented, the predisposition toward CNS progression during crizotinib is attributable to poor accumulation of the drug into the CNS.Citation46–Citation49
Next-generation ALK inhibitors have been developed to be more potent and with improved BBB penetration compared to crizotinib, and this has resulted in significant intracranial activity and CNS disease control. In Phase I and Phase II studies in crizotinib-resistant patients with measurable lesions at baseline, the reported ORR and intracranial DCR were 36%–45% and 80% for ceritinib,Citation72,Citation74 64% and 90% for alectinib (in the pooled analysis of NP28761 and NP28673 studies)Citation79 and 53% and 86% for brigantinib, respectively ().Citation80 In contrast to crizotinib and ceritinib, alectinib is not a P-gp substrate and can achieve higher CNS levels compared to these compounds, as demonstrated in preclinical models and also in clinical studies.Citation71,Citation81 Also, preliminary clinical results with brigatinib confirm its significant CNS activity demonstrated in preclinical studies in an orthotopic mouse brain tumor model.Citation82 Another potent, next-generation ALK and ROS1 inhibitor, lorlatinib, has shown preclinical and clinical intracranial activity.Citation83–Citation85 Indeed, lorlatinib induced regression of EML4–ALK-driven brain metastases, leading to prolonged survival in vivo. In an ongoing Phase I study, lorlatinib was associated with an intracranial ORR on target lesions of 42% in ALK-positive patients and an impressive intracranial ORR of 80% in ROS1-positive patients.Citation85
The ASCEND-7 is a multicenter, open-label, Phase II ongoing study to evaluate prospectively the efficacy and safety of ceritinib (administered orally once daily at a dose of 750 mg) in patients with ALK-positive NSCLC, metastatic to the brain without evidence of leptomeningeal carcinomatosis (previously treated or untreated with radiation to the brain and with or without prior exposure to crizotinib), and in patients with leptomeningeal carcinomatosis with or without evidence of active lesion at the baseline gadolinium-enhanced brain MRI (previous treatment with ALK inhibitors other than crizotinib is not allowed in the latter arm). Overall response rate is the primary outcome. Secondary outcome measures include time to intracranial tumor response, PFS and OS (NCT02336451).
Safety and tolerability
Ceritinib was well tolerated in all clinical studies, with most AEs of grade 1 and 2 and very rare events of grade ≥3 (). In the dose-escalation phase of the ASCEND-1, in which ceritinib was administered at dose levels of 50–750 mg daily, dose-limiting toxicities occurred in six patients at doses of ≥400 mg and included nausea and vomiting at 750 mg/daily, diarrhea at a daily dose ≥600 mg, dehydration at 600 mg daily, increase in alanine aminotransferase (ALT) and hypophosphatemia at 400 mg daily.Citation68 The most common AEs of any grade occurring among the 130 patients included in the study were nausea (82%), diarrhea (47%), vomiting (65%), fatigue (47%) and increased ALT levels (35%). The most common AEs of grade 3 or 4 related to the study drug were increased ALT levels (21%), increased aspartate aminotransferase levels (11%), diarrhea (7%) and increased lipase levels (7%); other less-common grade 3 or 4 toxicities included hypophosphatemia (3%), elevated amylase level and hyperglycemia (both 2%). No treatment-related deaths were reported. All toxicities were reversible on discontinuation of treatment. However, a high percentage of patients required dose reduction (51% of patients treated across all dose levels and 62% of patients treated with 750 mg daily). Four cases of interstitial lung disease and one case of asymptomatic grade 3 prolongation of the corrected QT interval were reported, possibly related to ceritinib therapy. In 8 (6%) of 130 patients, ceritinib treatment was permanently discontinued. In the update analysis of ASCEND-1, including patients with ALK-rearranged NSCLC who received ceritinib at 750 mg/day, 97% of patients had AEs suspected to be related to treatment.Citation72 The most common AEs of grade 1–2 were gastrointestinal disorders (diarrhea, nausea and vomiting), which occurred in 99% of patients and were manageable through use of concomitant medication and dose modifications. The most common grade 3–4 AEs (nonlaboratory and laboratory) were diarrhea and nausea (both 6%), increased ALT (30%) and aspartate aminotransferase (10%), increased lipase (7%) and hyperglycemia (6%; 2% was reported as an SAE), as shown in . Grade 3 or 4 treatment-related AEs and SAEs of any grade were reported in 51% and 12% of patients, respectively. Interstitial lung disease or pneumonitis was reported in nine (4%) patients (grade ≥3 in eight patients, including one leading to death), and no case of prolongation of the corrected QT interval (>500 ms) was observed. Twenty-six (11%) patients discontinued treatment due to AEs.
Safety analyses from the ASCEND-2 were consistent with those of ASCEND-1, with no new or unexpected SAE.Citation74 Gastrointestinal disorders, mainly of grade 1 or 2 (nausea 81.4%, diarrhea 80% and vomiting 62.9%), were the most prevalent AEs. Overall, grade ≥3 AEs were reported in 71.4% of all patients, and in 45.7% of patients, they were suspected to be drug related (). Among these, the ALT and γ-GT increase occurred in 15.7% and 9.3% of patients, respectively. Prolongation of QTc and pneumoni-tis occurred at any grade in 1.4% and 7.9% of patients, respectively, and grade ≥3 of these AEs each occurred in one patient. Drug-related SAEs were reported in 17.1% of patients. Only 7.9% of patients discontinued treatment due to an AE.Citation74 PROs showed a trend toward improved symptom burden and QoL was maintained during treatment. Also, in the ASCEND-3 trial, ceritinib was associated with pre-dominantly gastrointestinal AEs, mostly of grade 1 and 2, and they were manageable with dose interruptions and/or reduction.Citation75 Grade ≥3 AEs occurred in 106 (85.5%) of 124 patients and the most common AEs were increases in ALT and γ-GT (). Drug-related SAEs were observed in 11% of patients and ~80% of patients had dose interruptions or adjustments due to AEs. Among the 10 deaths, only 3 were due to AEs, none of which was considered drug related. Updated PRO results were consistent with the primary analysis and showed improvement in symptoms from baseline, and the QoL was maintained.
Phase III studies, ASCEND-4Citation76 and ASCEND-5,Citation77 confirmed the overall manageable safety profile of ceritinib (), with higher frequency of dose interruptions and modifications due to AEs compared to chemotherapy, but lower rates of treatment discontinuation. In both studies, ceritinib improved the lung cancer symptoms and the QoL, compared to chemotherapy.
Acquired resistance to ceritinib
Ceritinib has been widely demonstrated to have high activity against the most common mutations conferring resistance to crizotinib, including L1196M, G1269A and S1206Y.Citation67 Ceritinib has also activity against cell lines with secondary I1171T and V1180L mutations, conferring resistance to both crizotinib and alectinib.Citation86,Citation87 However, as observed for crizotinib and other targeted therapies, ALK-positive cells can acquire resistance under chronic exposure to ceritinib, thus determining disease progression and limiting its long-term efficacy. Molecular profiling of tumor tissues from rebiopsies obtained at the time of disease progression on ceritinib revealed the presence of novel, acquired mutations at either G1202 or F1174, in 5 of 11 samples analyzed.Citation67 Moreover, two different ceritinib resistance mutations were identified within two different biopsy sites in the same patient, underscoring the heterogeneity of ceritinib resistance mechanisms. As commented, the mutation G1202R maps to the solvent-exposed region of ALK, where the bulkier, charged side chain is thought to lead to steric hindrance of most ALK inhibitors. In the ASCEND-1 study, a patient who was found with G1202R did not respond to ceritinib.Citation73 The presence of this mutation confers resistance to ceritinib, while F1174C/L mutations map adjacent to the C terminus of the αC helix and may stabilize an active conformation that increases the ATP-binding affinity of ALK. Toyokawa et al identified an ALK G1123S mutation in a patient with ALK-positive NSCLC acquiring resistance to ceritinib. Such a mutation located at codon 1123, within the glycine-rich loop, seems to sterically block ATP binding and/or alter the dynamics of the glycine-rich loop, resulting in perturbation of the interactions with ALK inhibitors.Citation88
New fundamental insights about the mechanisms of resistance developing during treatment with different ALK inhibitors, including ceritinib, have been provided by a recent in-depth molecular characterization of a large series of repeat postprogression biopsies from ALK-positive NSCLC patients.Citation89 Overall, ALK resistance mutations represented the predominant mechanism of resistance after progression to next-generation ALK inhibitors; they were present in 56% of tumor samples, with distinct frequency and patterns of mutations depending on the drug, although the most common was represented by ALK G1202R. Among the 24 ceritinib-resistant samples, 54% showed resistance mutations, the most common being G1202R and F1174C/L (). Other mutations included C1156Y, V1180L and the novel G1202del.Citation89 Also, an EMT phenotype was identified in 5 of 12 (42%) ceritinib-resistant biopsy specimens, including 2 cases with the L1196M mutation which does not usually confer resistance to ceritinib.Citation89 Interestingly, the potent third-generation ALK inhibitor, lorlatinib, inhibited the growth of ceritinib-resistant patient-derived cell lines harboring ALK-resistant mutations, while it had no activity against ALK wild-type cell lines. In addition, lorlatinib was active against cell lines with compound mutations which can occur after sequential treatment with different ALK inhibitors.Citation89 Activation of this bypass track has emerged as a possible mechanism of acquired resistance to ceritinib. Indeed, in a tumor sample obtained from a patient after ceritinib progression, a MET amplification was identified.Citation73 A recent in vitro study investigated the mechanisms of acquired resistance to alectinib and ceritinib in H3122 NSCLC cell lines harboring the EML4–ALK variant 1 fusion.Citation90 Secondary mutations of ALK were not detected; in contrast, overexpression of phospho-ALK and alternative RTKs such as phospho-EGFR, phospho-HER3 and phospho-IGFR-1R was observed in these resistant cell lines. Furthermore, neuregulin 1 (NRG1), a ligand for HER3, was found to be upregulated and responsible for resistance by activating the EGFR family pathways through the NRG1–HER3–EGFR axis.Citation90
Finally, P-gp overexpression mediated resistance in ALK-rearranged NSCLC patients who received ceritinib or crizotinib therapy, but not alectinib.Citation91 Such resistance occurs independent of the presence of ALK mutations and other major activated oncogenes. Indeed, it was demonstrated that brain penetration of ceritinib is strongly restricted by Abcb1a/1b on BBB and by Abcg2 only in the absence of Abcb1a/1b.Citation71 Thus, the determination of P-gp expression may provide additional useful information to select the optimal ALK TKI therapy.
Ongoing studies and therapeutic combinations
After crizotinib, ceritinib was the second ALK inhibitor to receive approval, thus expanding the array of effective therapeutic options for ALK-positive NSCLC. However, there is still room for improvement and numerous ongoing trials are investigating whether combinatorial strategies can be more effective than ceritinib as a single agent for improving the outcome of ALK-rearranged NSCLC patients (). Heat shock protein 90 (Hsp90) is a chaperone protein that regulates the folding, stabilization and function of not only different client proteins, including the growth factors and signaling molecules, but also of oncogenic kinases including EGFR, BRAF, c-MET and EML4/ALK.Citation92 Preclinical data demonstrated that Hsp90 inhibitors were effective against cell lines and xenograft models positive for the EML4–ALK fusion gene. In ALK-driven NSCLC cells and xenografts of NSCLC resistant to ALK inhibitors, superior antitumor efficacy was observed when combining the Hsp90 inhibitor ganetespib with other targeted ALK agents.Citation93 Ganetespib overcame crizotinib resistance due to multiple forms of resistance, including ALK secondary mutations. In a Phase II trial of ganetespib in advanced NSCLC patients, in a cohort of 23 patients with NSCLC, wild type for both EGFR and KRAS, ALK rearrangements were found in 8 patients. Of these, four had partial response and three had stable disease.Citation94 Another potent HSP90 inhibitor, luminespib (AUY922), demonstrated significant activity in crizotinib-naïve and crizotinib-resistant patients in an initial Phase II trial, with the most common all grade toxicities being eye disorders, diarrhea and nausea.Citation95 However, in another more recent Phase II trial of luminespib monotherapy in ALK-positive patients who had progressed on prior ALK TKIs, no objective responses were observed among the six patients enrolled. Despite the low number of patients and premature closure of the study, these results suggest that monotherapy with this HSP90 inhibitor may not be a valid treatment option in this setting.Citation96 A Phase Ib, open-label, dose escalation study evaluating the combination of ceritinib and luminespib in crizotinib-resistant, ALK-rearranged NSCLC patients (NCT01772797) has been completed and its results are pending ().
Table 3 Summary of selected ongoing trials of ceritinib (LDK378) in NSCLC
Other possible therapeutic combinations to overcome or delay resistance to ceritinib are suggested by in vitro studies, such as the potential of combining the irreversible EGFR TKI, afatinib, with ceritinib in those cases of resistance mediated by activation of EGFR family pathways.Citation90
A Phase Ib/II study of the ALK inhibitor ceritinib in combination with the CDK4/6 inhibitor LEE011 (ribociclib) in patients with ALK-positive NSCLC is currently recruiting participants. The purpose of this study is to determine the MTD/recommended phase 2 dose (RP2D) of ribociclib and ceritinib combination and to evaluate whether this combination is safe and has activity in ALK-positive advanced NSCLC patients, who are ALK-inhibitor-naïve or have progressed after treatment with an ALK inhibitor other than ceritinib or after treatment with ceritinib (NCT02292550). The introduction of immune checkpoint inhibitors, including monoclonal antibodies directed against the cytotoxic T-lymphocyte-associated antigen-4 and the programmed cell death protein-1/programmed cell death ligand-1 (PD-1/PD-L1) pathways, has improved the treatment of advanced NSCLC, especially for those patients without targetable activated oncogenes.Citation97 There is strong scientific rationale for sequentially or concurrently combining ALK inhibitors with immune checkpoint blockade.Citation45,Citation98,Citation99 However, recent data from clinical trials suggest that anti-PD-1/PD-L1 antibodies are less effective in never-smokers, which is the population enriched for EGFR mutations or ALK rearrangements, compared to smokers, probably for increased tumor immunogenicity due to a higher mutational load in smoking-associated lung cancer.Citation97 Moreover, poor responses to PD-1/PD-L1 inhibitors have been observed in EGFR-mutated or ALK-rearranged NSCLC patients and this can be explained by low rates of concurrent PD-L1 expression and CD8(+) tumor-infiltrating lymphocytes (TILs) within the tumor microenvironment.Citation100 Some ongoing studies are evaluating the combination of ALK inhibitors and checkpoint inhibitors, including an open-label, multicenter, Phase I trial of ceritinib in combination with the anti-PD1 monoclonal antibody, nivolumab, in ALK-positive, stage IIIB or IV NSCLC. Primary outcome measures are MTD and/or recommended dose for the expansion phase and ORR. Secondary outcome measures include DOR, DCR and OS (NCT02393625). We should mention that molecular docking simulations and preclinical studies demonstrate that ceritinib may also be active against ROS1-rearranged NSCLC and some clinical reports confirm this activity.Citation66,Citation101,Citation102 Ongoing studies are evaluating the role of ceritinib in this subset of patients ().
Discussion and conclusion
Since the approval of the first-in-class ALK inhibitor, crizo-tinib, in 2011, the landscape of treatment for ALK-positive NSCLC has rapidly evolved over the last few years, thus leading to a significant improvement in the prognosis of this molecularly defined subset of NSCLC patients.
Crizotinib was approved based on pronounced clinical benefit observed in early-phase studies in ALK-rearranged NSCLC patients. However, despite its remarkable activity, the vast majority of patients inevitably progress due to acquired resistance, with the brain being a common site of relapse. The most common mechanisms underlying acquired resistance have been characterized, and these include ALK secondary mutations or copy number alterations and activation of alternative bypass signaling pathways. In order to face the major shortcomings of crizotinib, various next-generation ALK inhibitors have been developed and are currently at different phases of clinical development. These are structurally distinct, have higher potency against ALK and ALK with resistance mutations and improved BBB penetration, compared to crizotinib. Ceritinib, a highly potent and selective ALK inhibitor, was the first to be approved by the FDA in 2014 for the treatment of ALK-positive NSCLC patients with progression, or who are intolerant to crizotinib, based on pronounced clinical activity observed in the Phase I ASCEND-1 trial, and it is currently approved in the European Union and in several countries worldwide with this indication. In Phase I and II studies, ceritinib treatment produced significant objective responses in both crizotinib-resistant and crizotinib-naïve ALK-positive NSCLC patients, and recent results from Phase III studies have demonstrated that ceritinib significantly improves PFS compared with standard chemotherapy in the first- and second-line treatment setting of ALK-positive, advanced NSCLC patients, with associated significant improvements in QoL and lung cancer symptoms. Thus, this great amount of data suggests that ceritinib is a solid therapeutic choice in crizotinib-resistant ALK-positive NSCLC patients, including those with CNS involvement. Sequential crizotinib and ceritinib in a multi-institutional retrospective analysis was associated with a PFS of 17.4 months and an impressive OS of 49.4 months, suggesting that sequential use of different ALK inhibitors is an effective strategy in ALK-positive NSCLC patients.Citation103 Despite the success of ceritinib in clinical development, other available next-generation inhibitors have shown robust clinical activity in crizotinib-pretreated patients, including alectinib, which has already been approved by the FDA and the EMA, and brigatinib, which has received breakthrough therapy designation by the FDA. Notably, the activity of these next-generation ALK inhibitors has been also observed in crizotinib-naïve patients, and alectinib and brigatinib are currently being investigated in Phase III studies in a head-to-head comparison with crizotinib in ALK-inhibitor-naïve patients.
Therefore, since ceritinib has gained an unquestionable role in the therapeutic algorithm of ALK-positive NSCLC patients, with the availability of new and active next-generation ALK inhibitors, selecting the appropriate sequence of ALK TKIs has become a crucial factor to improve patient outcomes. After crizotinib failure, some important points can be helpful in guiding selection between the two clinically approved next-generation ALK inhibitors, ceritinib or alectinib. One is the intracranial activity of these compounds. Indeed, alectinib is not a P-gp substrate and can achieve higher CNS levels compared to ceritinib or crizotinib, thereby improving CNS disease control, as demonstrated in clinical studies.Citation71,Citation79,Citation81 Therefore, alectinib can be considered a preferred option for ALK-positive NSCLC patients with CNS progression. Also, the side effect profile is slightly different between ceritinib and alectinib, with clinical data suggesting alectinib is overall better tolerated than ceritinib. Recent data suggest that treatment choice at progression should be guided by underlying resistance mutations. Indeed, ceritinib is not only able to overcome the most common crizotinib-resistant mutations, but also has activity against some mutations conferring resistance to alectinib.Citation67,Citation86,Citation87 However, acquired resistance to ceritinib may eventually occur, with secondary ALK mutations being the most common mechanism of resistance found in postprogression patient samples. G1202R and F1174C/L represent the predominant resistance mutations after ceritinib treatment.Citation89 The F1174 mutation confers resistance to ceritinib, but is still sensitive to alectinib, while the G1202R is resistant to most next-generation ALK inhibitors, except lorlatinib. This potent third-generation ALK inhibitor was able to inhibit the growth of ceritinib-resistant patient-derived cell lines harboring ALK resistance mutations, including G1202R, and was also active against cell lines with compound mutations, which occur when patients are sequentially treated with different ALK TKIs.Citation89 Of note, in a recent report, a secondary mutation conferring resistance to lorlatinib was identified, and this mutation, ALK L1198F, paradoxically enhanced binding to crizotinib, thus resensitizing resistant cancers to crizotinib.Citation104 Overall, these data corroborate the use of molecular profiling at disease progression to match the treatment choice to each acquired mutation. In those cases in which resistance to ceritinib is associated with ALK-independent mechanisms, such as activation of alternative kinases involved in proliferation signaling pathways or epithelial–mesenchymal transition (EMT), rational combinations of drugs can repre-sent a promising therapeutic option, as suggested by some preclinical evidence. Some studies are currently evaluating combinations of immune checkpoint inhibitors with ALK inhibitors, and results in terms of activity and toxicity of these novel combinations are also eagerly awaited.
Disclosure
The authors report no conflicts of interest in this work.
References
- FerlayJSoerjomataramIDikshitRCancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012Int J Cancer20151365 E359 E38625220842
- SandlerAGrayRPerryMCPaclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancerNEJM200635524 2542 255017167137
- ReckMvon PawelJZatloukalPPhase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAilJ Clin Oncol2009278 1227 123419188680
- ThatcherNHirschFRLuftAVSQUIRE InvestigatorsNecitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trialLancet Oncol2015167 763 77426045340
- PolitiKHerbstRSLung cancer in the era of precision medicineClin Cancer Res20152110 2213 222025979927
- SantarpiaMAltavillaGSalazarMFTyrosine kinase inhibitors for non-small-cell lung cancer: finding patients who will be responsiveExpert Rev Respir Med201153 413 42421702662
- RielyGJYuHAEGFR: the paradigm of an oncogene-driven lung cancerClin Cancer Res20152110 2221 222625979928
- KatayamaRLovlyCMShawATTherapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicineClin Cancer Res20152110 2227 223525979929
- MastersGATeminSAzzoliCGSystemic therapy for stage IV non-small-cell lung cancer: American Society of Clinical Oncology Clinical Practice Guideline UpdateJ Clin Oncol20153330 3488 351526324367
- SodaMChoiYLEnomotoMIdentification of the transforming EML4-ALK fusion gene in non-small-cell lung cancerNature20074487153 561 56617625570
- IwaharaTFujimotoTWenDMolecular characterization of ALK, a receptor tyrosine kinase espressed specifically in the nervous systemOncogene1997144 439 4499053841
- ChiarleRVoenaCAmbrogioCPivaRInghiramiGThe anaplastic lymphoma kinase in the pathogenesis of cancerNat Rev Cancer200881 11 2318097461
- PalmeriRHVernerssonEGrabbeCHallbergBAnaplastic lymphoma kinase: signalling in development and diseaseBiochem J20094203 345 36119459784
- StoicaGEKuoAAignerAIdentification of anaplastic lymphoma kinase as receptor for the growth factor pleiotrophinJ Biol Chem200127620 16772 1677911278720
- StoicaGEKuoAPowersCMidkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell typesJ Biol Chem200227739 35990 3599812122009
- HallbergBPalmerRHMechanistic insight into ALK receptor tyrosine kinase in human cancer biologyNat Rev Cancer20131310 685 70024060861
- SodaMTakadaSTakeuchiKA mouse model for EML4-ALK-positive lung cancerProc Natl Acad Sci U S A200810550 19893 1989719064915
- McDermottUIafrateAJGrayNSGenomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitorsCancer Res2008689 3389 339518451166
- SasakiTRodigSJChirieacLRThe biology and treatment of EML4-ALK non-small cell lung cancerEur J Cancer20104610 1773 178020418096
- ChoiYLTakeuchiKSodaMIdentification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancerCancer Res20086813 4971 497618593892
- KoivunenJPMermelCZejnullahuKEML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancerClin Cancer Res20081413 4275 428318594010
- GalkinAVMelnickJSKimSIdentification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALKProc Natl Acad Sci U S A20071041 270 27517185414
- HeuckmannJMBalke-WantHMalchersFDifferential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variantsClin Cancer Res20121817 4682 469022912387
- YoshidaTOyaYTanakaKDifferential crizotinib response duration among ALK fusion variants in ALK-positive non-small-cell lung cancerJ Clin Oncol20163428 3383 338927354483
- RikovaKGuoAZengQGlobal survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancerCell20071316 1190 120318083107
- TakeuchiKChoiYLTogashiYKIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancerClin Cancer Res2009159 3143 314919383809
- TogashiYSodaMSakataSKLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue onlyPLoS One201272 e3132322347464
- ChoiYLLiraMEHongMA novel fusion of TPR and ALK in lung adenocarcinomaJ Thorac Oncol201494 563 56624736082
- OuSHKlempnerSJGreenboweJRIdentification of a novel HIP1-ALK fusion variant in non-small-cell lung cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to alectinibJ Thorac Oncol2014912 1821 182525393796
- ShanLJiangPXuFBIRC6-ALK, a novel fusion gene in ALK break-apart FISH-negative lung adenocarcinoma, responds to crizotinibJ Thorac Oncol201510 e37 e3926001147
- ShawATYeapBYMino-KenudsonMClinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALKJ Clin Oncol20092726 4247 425319667264
- LazzariCSpitaleriGCataniaCTargeting ALK in patients with advanced non small cell lung cancer: biology, diagnostic and therapeutic optionsCrit Rev Oncol Hematol2014893 358 36524156959
- GainorJFVargheseAMOuSHALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancerClin Cancer Res20131915 4273 428123729361
- Pekar-ZlotinMHirschFRSoussan-GutmanLFluorescence in situ hybridization, immunohistochemistry, and next-generation sequencing for detection of EML4-ALK rearrangement in lung cancerOncologist2015203 316 32225721120
- WynesMWShollLMDietelMAn international interpretation study using the ALK IHC antibody D5F3 and a sensitive detection kit demonstrates high concordance between ALK IHC and ALK FISH and between evaluatorsJ Thorac Oncol201495 631 63824722153
- RogersTMRussellPAWrightGComparison of methods in the detection of ALK and ROS1 rearrangements in lung cancerJ Thorac Oncol2015104 611 61825789833
- MarchettiADi LoritoAPaceMVALK protein analysis by IHC staining after recent regulatory changes: a comparison of two widely used approaches, revision of the literature, and a new testing algorithmJ Thorac Oncol2016114 487 49526916631
- ChristensenJGZouHYArangoMECytoreductive antitumor activity of PF 2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphomaMol Cancer Ther2007612 Pt 1 3314 332218089725
- CuiJJTran-DubéMShenHStructure based drug design of crizotinib (PF 02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK)J Med Chem20115418 6342 636321812414
- KwakELBangYJCamidgeDRAnaplastic lymphoma kinase inhibition in non-small-cell lung cancerN Engl J Med201036318 1693 170320979469
- CamidgeDRBangYJKwakELActivity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 studyLancet Oncol20121310 1011 101922954507
- KimDWAhnMJYangPCUpdated results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC)Ann Oncol201223Suppl 9 ix400 ix446
- ShawATKimDWNakagawaKCrizotinib versus chemotherapy in advanced ALK-positive lung cancerN Engl J Med201336825 2385 239423724913
- SolomonBJMokTKimDWFirst-line crizotinib versus chemotherapy in ALK-positive lung cancerN Engl J Med201437123 2167 217725470694
- SantarpiaMGilNRosellRStrategies to overcome resistance to tyrosine kinase inhibitors in non-small-cell lung cancerExpert Rev Clin Pharmacol201584 461 47726068305
- CostaDBKobayashiSPandyaSSCSF concentration of the anaplastic lymphoma kinase inhibitor crizotinibJ Clin Oncol20112915 e443 e44521422405
- ChunSGChoeKSIyengarPYordyJSTimmermanRDIsolated central nervous system progression on Crizotinib: an Achilles heel of non-small cell lung cancer with EML4-ALK translocation?Cancer Biol Ther20121314 1376 138322986231
- TangSCNguyenLNSparidansRWWagenaarEBeijnenJHSchinkelAHIncreased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridarInt J Cancer20141346 1484 149424037730
- Dagogo-JackIShawATCrizotinib resistance: implications for therapeutic strategiesAnn Oncol201627Suppl 3 iii42 iii5027573756
- DoebeleRCPillingABAisnerDLMechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancerClin Cancer Res2012185 1472 148222235099
- KatayamaRShawATKhanTMMechanisms of acquired crizotinib resistance in ALK-rearranged lung cancersSci Transl Med20124120 120ra17
- ChoiYLSodaMYamashitaYEML4-ALK mutations in lung cancer that confer resistance to ALK inhibitorsN Engl J Med201036318 1734 173920979473
- SasakiTKoivunenJOginoAA novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitorsCancer Res201118 6051 606021791641
- ToyokawaGHiraiFInamasuESecondary mutations at I1171 in the ALK gene confer resistance to both Crizotinib and AlectinibJ Thorac Oncol2014912 e86 e8725393798
- OuSHAzadaMHsiangDJNext-generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinibJ Thorac Oncol201494 549 55324736079
- KimSKimTMKimDWHeterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancerJ Thorac Oncol201384 415 42223344087
- TanizakiJOkamotoIOkabeTActivation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancerClin Cancer Res201218 6219 622622843788
- KimuraMEndoMInoueTAnalysis of ERBB ligand-induced resistance mechanism to crizotinib by primary culture of lung adenocarcinoma with EML4-ALK fusion geneJ Thorac Oncol2015103 527 53025695223
- LovlyCMMcDonaldNTChenHRationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancerNat Med2014209 1027 103425173427
- CrystalASShawATSequistLVPatient-derived models of acquired resistance can identify effective drug combinations for cancerScience20143466216 1480 148625394791
- HrustanovicGOlivasVPazarentzosERAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancerNat Med2015219 1038 104726301689
- GalkinAVMelnickJSKimSIdentification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALKProc Natl Acad Sci U S A20071041 270 27517185414
- MarsiljeTHPeiWChenBSynthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5 chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2 (isopropylsulfonyl)phenyl) pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trialsJ Med Chem20135614 5675 569023742252
- ChenJJiangCWangSLDK378: a promising anaplastic lymphoma kinase (ALK) inhibitorJ Med Chem20135614 5673 567423837797
- ShawATMehraRKimDWClinical activity of the ALK Inhibitor LDK378 in advanced, ALK-positive NSCLPresented at the Annual Meeting of the American Society of Clinical OncologyMay 31–June 4, 2013Chicago Abstract
- ZYKADIA™ (ceritinib) capsules, for oral use: US prescribing information2014 Available from: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdfAccessed March 1, 2017
- FribouletLLiNKatayamaRThe ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancerCancer Discov201446 662 67324675041
- ShawAKimDMehraRCeritinib in ALK-rearranged non-small-cell lung cancerN Engl J Med201437013 1189 119724670165
- LauYYGuWLinTSongDYuRScottJWEffects of meal type on the oral bioavailability of the ALK inhibitor ceritinib in healthy adult subjectsJ Clin Pharmacol2016565 559 56626272586
- KhozinSBlumenthalGMZhangLFDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancerClin Cancer Res20152111 2436 243925754348
- KortASparidansRWWagenaarEBeijnenJHSchinkelAHBrain accumulation of the EML4-ALK inhibitor ceritinib is restricted by P-glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2)Pharmacol Res2015102 200 20726361725
- KimDWMehraRTanDSActivity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trialLancet Oncol2016174 452 46326973324
- TanDSKimDWThomasMGenetic landscape of ALK+ non-small cell lung cancer (NSCLC) patients (pts) and response to ceritinib in ASCEND-1J Clin Oncol201634 (suppl; abstr 9064); ASCO Meet Abstr
- CrinòLAhnMJDe MarinisFMulticenter Phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2J Clin Oncol20163424 2866 287327432917
- FelipEOrlovSParkKPhase 2 study of ceritinib in ALKi-naïve patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC): Whole body responses in the overall pt group and in pts with baseline brain metastases (BM)Presented at: The ESMO Congress 2016 (abstr 1208O)October 7–11; 2016Copenhagen, Denmark
- SoriaJCTanDSChiariRFirst-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 studyLancet201738910072 917 92928126333
- ScagliottiGKimTCrinòLCeritinib versus chemotherapy (CT) in patients (pts) with advanced anaplastic lymphoma kinase (ALK)-rearranged (ALK+) non-small cell lung cancer (NSCLC) previously treated with CT and crizotinib (CRZ): Results from the confirmatory phase 3 ASCEND-5 studyPresented at the ESMO Congress 2016 (abstr LBA42_PR)October 7–11; 2016Copenhagen, Denmark
- CostaDBShawATOuSHClinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastasesJ Clin Oncol2015103317 1881 188825624436
- GadgeelSMShawATGovindanRPooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small-cell lung cancerJ Clin Oncol20163434 4079 408527863201
- GettingerSNBazhenovaLALangerCJActivity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trialLancet Oncol20161712 1683 169627836716
- SantarpiaMAltavillaGRosellRAlectinib a selective, next-generation ALK inhibitor for treatment of ALK-rearranged non-small-cell lung cancerExpert Rev Respir Med201593 255 26825652176
- ZhangSAnjumRSquillaceRThe potent ALK inhibitor brigatinib (AP26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical modelsClin Cancer Res20162222 5527 553827780853
- JohnsonTWRichardsonPFBaileySDiscovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutationsJ Med Chem20145711 4720 474424819116
- ZouHYFribouletLKodackDPPF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical modelsCancer Cell2015281 70 8126144315
- FelipEBauerTMSolomonBSafety and Efficacy of Lorlatinib (PF-06463922) in Patients with Advanced ALK+ or ROS1+ Non-Small-Cell Lung Cancer (NSCLC)Presented at the IASLC 17th World Congress on Lung Cancer (abstr ID 5053)December 4–7, 2016Vienna, Austria
- ToyokawaGHiraiFInamasuESecondary mutations at I1171 in the ALK gene confer resistance to both crizotinib and alectinibJ Thorac Oncol2014912 e86 e8725393798
- KatayamaRFribouletLKoikeSTwo novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinibClin Cancer Res20142022 5686 569625228534
- ToyokawaGInamasuEShimamatsuSIdentification of a novel ALK G1123S mutation in a patient with ALK-rearranged non–small-cell lung cancer exhibiting resistance to ceritinibJ Thorac Oncol2015107 e55 e5726134233
- GainorJFDardaeiLYodaSMolecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancerCancer Discov2016610 1118 113327432227
- DongXFernandez-SalasELiEWangSElucidation of resistance mechanisms to second-generation ALK inhibitors alectinib and ceritinib in non-small cell lung cancer cellsNeoplasia2016183 162 17126992917
- KatayamaRSakashitaTYanagitaniNP-glycoprotein mediates ceritinib resistance in anaplastic lymphoma kinase-rearranged non-small cell lung cancerEBioMedicine20163 54 6626870817
- TrepelJMollapourMGiacconeGNeckersLTargeting the dynamic HSP90 complex in cancerNat Rev Cancer2010108 537 54920651736
- ChenZSasakiTTanXInhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogeneCancer Res20107023 9827 983620952506
- SocinskiMAGoldmanJEl-HariryIA multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancerClin Cancer Res20131911 3068 307723553849
- FelipECarcerenyEBarlesiFPhase II activity of the Hsp90 inhibitor AUY922 in patients with ALK-rearranged (ALK+) or EGFRmutated advanced non-small cell lung cancer (NSCLC)Ann Oncol201223Suppl 9 ix152 ix174 438
- GainorJFMarcouxJPRabinMA phase II trial of AUY922, a heat shock protein 90 (HSP90) inhibitor, in ALK-positive lung cancer patients previously treated with ALK inhibitorsJ Thorac Oncol201510Suppl 2 S66 S89026710300
- SantarpiaMGiovannettiERolfoCRecent developments in the use of immunotherapy in non-small cell lung cancerExpert Rev Respir Med2016107 781 79827148808
- VannemanMDranoffGCombining immunotherapy and targeted therapies in cancer treatmentNat Rev Cancer2012124 237 25122437869
- OtaKAzumaKKawaharaAInduction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancerClin Cancer Res20152117 4014 402126019170
- GainorJFShawATSequistLVEGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysisClin Cancer Res20162218 4585 459327225694
- DavareMAVelloreNAWagnerJPStructural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitorsProc Natl Acad Sci U S A201511239 E5381 E539026372962
- SubbiahVHongDSMeric-BernstamaFClinical activity of ceritinib in ROS1-rearranged non-small cell lung cancer: bench to bedside reportProc Natl Acad Sci U S A201611311 E1419 E142026917690
- GainorJFTanDSDe PasTProgression-free and overall survival in ALK-positive NSCLC patients treated with sequential crizotinib and ceritinibClin Cancer Res20152112 2745 275225724526
- ShawATFribouletLLeshchinerIResensitization to crizotinib by the lorlatinib ALK resistance mutation L1198FN Engl J Med20163741 54 6126698910