
Abstract
Computer-aided drug discovery and development approaches such as virtual screening, molecular docking, and in silico drug property calculations have been utilized in this effort to discover new lead compounds against tuberculosis. The enzyme 7,8-diaminopelargonic acid aminotransferase (BioA) in Mycobacterium tuberculosis (Mtb), primarily involved in the lipid biosynthesis pathway, was chosen as the drug target due to the fact that humans are not capable of synthesizing biotin endogenously. The computational screening of 4.5 million compounds from the Enamine REAL database has ultimately yielded 45 high-scoring, high-affinity compounds with desirable in silico absorption, distribution, metabolism, excretion, and toxicity properties. Seventeen of the 45 compounds were subjected to bioactivity validation using the resazurin microtiter assay. Among the 4 actives, compound 7 ((Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl)cyclohexyl)amino)acetimidic acid) displayed inhibitory activity up to 83% at 10 μg/mL concentration against the growth of the Mtb H37Ra strain.
Introduction
The traditional process of drug discovery and development is generally viewed as extremely time-consuming and labor-intensive. Typically, it takes about 10–15 years,Citation1 and about US $800 million is required to complete the entire drug discovery and development timeline.Citation2 This traditional process generally involves the synthesis of huge compound libraries and high-throughput screenings to determine potential compounds with bioactivities.Citation3 However, this process has been proven to be quite unsuccessful, as hit rates were lowCitation4 and approximately 50% of the late-stage failures are attributed to unsatisfactory absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties.Citation5 Computer-aided drug discovery and development (CADDD) helps in reducing the time and cost of drug discovery by applying computational power that can consequently streamline the whole process.Citation6
CADDD can be applied to a variety of diseases, especially to those that cause major public health concerns and require immediate development of effective and safe treatments. For example, one serious malady that is afflicting one-third of the world population is tuberculosis or TB,Citation7 a respiratory disease caused by the bacterium Mycobacterium tuberculosis (Mtb). Hitherto, there is no concrete cure for this disease due to the continuing resurgence of drug-resistant strains, which calls for newer and more effective antitubercular drugs.
We have applied CADDD approaches in discovering possible leads against druggable targets in Mtb.Citation8–Citation17 An interesting drug target in Mtb is the enzyme 7,8-diaminopelargonic acid (DAPA) aminotransferase, more commonly known as Mtb BioA. BioA is involved in biotin biosynthesis pathway, one of the most important survival pathways of Mtb.Citation18 More specifically, BioA is the second enzyme in the pathway which catalyzes the conversion of 7-keto-aminopelargonic acid (KAPA) to DAPA.Citation19 Mtb is dependent on biotin synthesis for survival during latency. In the absence of biotin, disruption of the biotin synthesis pathway results in cell death rather than growth arrest.Citation20,Citation21 BioA has been implicated in long-term survival of mycobacteria,Citation22 more specifically for optimal stationary-phase growth in rich, biotin-replete medium.Citation23 Additionally, biotin synthesis is unique to plants and microorganisms.Citation24 The fact that humans lack this enzyme, as they are not capable of synthesizing biotin endogenously, makes it even more an ideal drug target for antitubercular drug discovery.Citation19
A known potent inhibitor of BioA is amiclenomycin (ACM),Citation25 specifically its cis isomer.Citation26 It is a mechanism-based inhibitor which works through covalent modification of the pyridoxal phosphate (PLP) cofactor of BioA via aromatization.Citation26,Citation27 Its simplified amino-alcohol analog, ACM-OH, is known to have better whole-cell activity against Mtb. Although ACM is an excellent inhibitor, it has poor chemical stability due to spontaneous aromatization,Citation26 which makes it a weak potential drug candidate. Thus, it is highly instructive to further search for compounds against BioA which may pave the way for the development of a new class of antituberculosis drugs.
In this study, we employed CADDD approaches in an effort to discover new leads against TB. Accordingly, Enamine REAL database was screened based on a pharmacophore generated from the active site of BioA. The high-scoring compounds were then subsequently docked into BioA’s active site, and the high-affinity hits were also subjected to in silico ADMET evaluation. The inhibitory activity of the compounds that passed the ADMET filter was then tested experimentally against Mtb H37Ra using the resazurin microtiter assay (REMA).
Materials and methods
Computational screening
All computational screening methods were performed using the Discovery Studio 4.0 (DS4.0; BIOVIA-Dassault Systèmes, formerly Accelrys) running on Windows 7 operating system in a machine with an Intel® Core™ i7-3770 3.40GHz quad core processor.
Generation of structure-based pharmacophore model
A three-dimensional (3D) crystal structure of DAPA synthase (BioA: Rv1568) (PDB ID: 3TFU)Citation27 was retrieved from the Protein Data Bank (www.rcsb.org).Citation28 The structure was then prepared and optimized using the Prepare Protein and Minimization protocols of DS4.0. Structure preparation such as insertion of missing atoms, optimization of side chain conformation, removal of alternate conformations, protonation of titratable residues, as well as modeling of missing loop regions was done in order to prime the enzyme target for further computational screening. After optimization, a binding-site sphere was subsequently defined on the enzyme around the location of the invariant residue, Lys283, which was covalently linked to the PLP cofactor.Citation20 Based on the chemical features (hydrophobic, H-donor, H-acceptor) of the generated binding-site sphere, a pharmacophore model was then generated by employing the Interaction Generation protocol of DS4.0.
Virtual screening of compound libraries
A total of 4.5 million compounds from the Enamine REAL database (parts 1–9) (http://www.enamine.net) were screened. By using the Prepare Ligands protocol, several conformations of the compounds were generated and were subsequently compiled into a single library using the Build 3D Database protocol of DS4.0. The ligand library was then screened against the pharmacophore model by rigid and flexible fitting methods run in succession. The high-scoring compounds (fit value scores >3.0) were subsequently subjected to molecular docking studies.
Molecular docking
Prior to docking the high-scoring compounds from the pharmacophore-based screening, validation of the CDOCKER docking protocol was performed by docking KAPA and ACM, known substrate and inhibitor of BioA, respectively. After confirming interaction profile reproducibility, the virtual screening hits were docked into the BioA active site. The binding affinity calculation was done using the Calculate Binding Energies protocol of DS4.0. The compounds with better binding energy values than ACM were subjected to another round of elimination using ADMET filters.
In silico ADMET prediction
The compounds selected for further screening were subjected to ADMET calculations. Parameters such as aqueous solubility, absorption, plasma protein binding, cytochrome P450 2D6 inhibition, and hepatotoxicity were all determined using the ADMET protocol in DS4.0 (). Moreover, the toxicity potential (ie, carcinogenicity and mutagenicity) of the compounds was also predicted using the TOPKAT (TOxicity Prediction by Komputer Assisted Technology) protocol in DS4.0 ().
Table 1 ADMET descriptor values in DS4.0 and their corresponding interpretations
Table 2 Binding energies, and ADMET predictions of ACM, and Enamine hit compounds
Protein ligand interaction diagram
Two-dimensional and 3D ligand interaction diagrams for the compounds that showed H37Ra growth-inhibitory activities, as well as that of KAPA, ACM, and ACM-OH, were generated in order to examine the pertinent amino acid interactions of these compounds with the protein active site.
Resazurin-based microtiter plate assay
Bacterial strains and culture conditions
Mtb H37Ra (ATCC 25177) bacterial stocks were provided by the Marine Natural Products Laboratory, Marine Science Institute, University of the Philippines. Bacterial stocks were thawed and plated on Middlebrook 7H11 agar supplemented with 10% oleic acid–albumin–dextrose–catalase (Titan Media, Delhi, India). Plates were then incubated at 37°C for 3–4 weeks, and then subsequently inoculated with Middlebrook 7H9 broth supplemented with 10% albumin–dextrose–catalase (Titan Media). Broth tubes were incubated in a shaking incubator at 37°C and 150 rpm for an additional 3–4 weeks.
Compound preparation
Hit compounds from the Enamine database were all procured from Enamine Ltd, Kiev, Ukraine. All compounds were solubilized in dimethyl sulfoxide (DMSO) at a stock concentration of 2 mg/mL, aliquoted, and stored at −20°C. Rifampicin (Sigma-Aldrich, St Louis, MO, USA) was used as the assay positive control.
REMA
A McFarland No 1 (A625 nm ≈0.25) culture of H37Ra was first prepared as the assay inoculum. The adjusted culture was then diluted further to a 1:49 mixture of culture:M7H9 broth. Test solutions of Enamine compounds and rifampicin were prepared in DMSO at final concentrations of 1 mg/mL (high) and 0.01 μg/mL (low). Since each test well had a final volume of 200 μL, and 2 μL of the compound was added per test well, the final well concentrations of the drug and rifampicin were 10 μg/mL and 0.1 μg/mL. Each sample was tested in quadruplicates.
The assay proper was performed in 96-well, flat-bottomed microtiter plates. The well plates were then incubated at 37°C and 150 rpm for 7 days in a shaking incubator. At day 7, 20 μL of 0.02% resazurin (Sigma-Aldrich) was added to all the wells and then incubated for an additional 24 hours. The fluorescence readings of all the wells were then read at an excitation filter of 530 nm and an emission filter of 590 nm.
Results and discussion
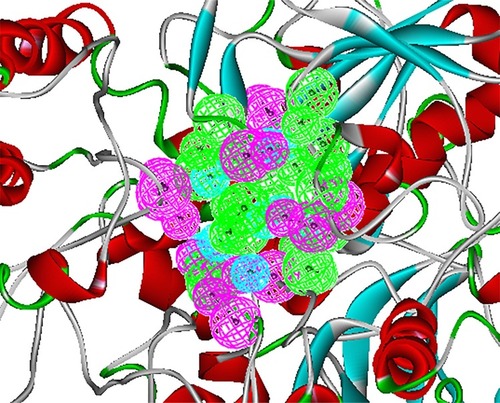
The crystal structure of BioA (PDB ID: 3TFU)Citation27 () was prepared and optimized prior to virtual screening experiment.Citation18 The optimization step, however, may impose changes in the conformation of the protein that might result in large deviations that can adversely affect the computational results. Delightfully, the root-mean-square deviation value of 0.70 Å, calculated via superimposition of the C-alpha pairs of the raw and the minimized protein structures, was highly satisfactory (). The binding-site sphere (x, y, z: 41.37, 6.1, −22.3; r=10 Å) was defined around the location of residue Lys283, which was covalently linked to the cofactor, PLP.Citation20 The pharmacophore model with 25 features (9 hydrophobic, 9 donors, and 7 acceptors; ) was the basis in the virtual screening of database ligands.
Figure 1 Preparation of 3D structure of Mycobacterium tuberculosis 7,8-diaminopelargonic acid aminotransferase: (A) 3D structure of 7,8-diaminopelargonic acid aminotransferase (BioA) of M. tuberculosis (LdtMt2, PDB ID: 3TFU)Citation27 (3D structure generated using: Protein Data Bank, www.rcsb.org);Citation28 (B) molecular overlay of raw (cyan) and minimized (yellow) crystal structures of BioA (RMSD =0.70 Å).
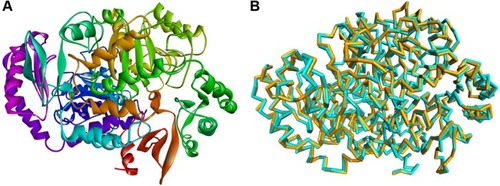
Figure 2 Structure-based pharmacophore model of BioA. This pharmacophore was modeled based on BioA’s active site, showing 9 hydrophobic (cyan), 9 donor (magenta), and 7 acceptor (green) features.
In this study, a total of 4.5 million compounds from the Enamine REAL database were prepared to remove duplicate structures, enumerate isomers and tautomers, and generate 3D conformations.Citation29 The Lipinski filter was turned off since a number of accounts showed that some compounds, including many natural products and natural product-like molecules, have become successful candidate drugs, despite violating the Lipinski’s Rule of Five.Citation30,Citation31 Virtual screening was done by rigid and flexible fitting methods with reference to a pharmacophore model generated based on the structure of the target. This step efficiently reduces the number of compounds that was included in the subsequent molecular docking studies.
To validate the applicability of the docking protocol, the reference compounds, KAPA and ACM, were docked in silico to BioA. As desired, the docked BioA–KAPA and BioA–ACM complexes involved interactions with similar residues as reported in previous studies.Citation20,Citation26 The van der Waals (vdW) interactions of KAPA with Tyr25, Trp64, and Phe402 were consistent with observations from the study of Dey et al.Citation20 In comparison to a more recently released crystal structure of BioA complexed with KAPA (PDB ID: 4CXQ),Citation32 the docked complex displayed similar hydrogen bonding between KAPA and Tyr157, and hydrophobic interaction with Trp64. The docked complex also predicted additional interactions that are not found in other studies, such as the charge–pi interaction with Trp64 and Phe402 of BioA, which are consistent with the experimental results observed by Dey et al.Citation20 However, the covalent bond formation between ACM and PLP has been represented as polar interaction in the docking results. The results also showed vdW forces with Tyr25, Tyr157, and Lys283, a conserved residue in BioA.Citation22
The docking experiment with ACM returned a binding energy of −158.81 kcal/mol, which was set as the threshold value in our screening. Out of over 4 million compounds, 45 turned out to have better binding energies than ACM, with acceptable predicted toxicity properties and favorable pharmacokinetic profiles. The calculated properties of 17 accessible compounds are detailed in . The mycobacterial activity of these hit compounds was subsequently tested in vitro using the H37Ra strain of Mtb.
In this study, the REMA was employed in whole-cell bioactivity measurements. Although target-based screenings usually employ inhibition assays against the target, whole-cell assay was utilized here because some compounds that have been active in target-based screens were often found to be inactive on the whole bacterium.Citation33,Citation34 REMA is considered as one of the most rapid, simple, yet inexpensive methods for drug susceptibility testing.Citation35 Moreover, since resazurin is a relatively inexpensive dye, the REMA can be feasibly applied in low-resource settings.Citation36 Out of 45 high-scoring, high-binding hits, only 17 () were available for acquisition and subjected to in vitro assay against the Mtb H37Ra strain. Each compound was tested at a high-dose concentration (10 μg/mL) and a low-dose concentration (0.1 μg/mL). The percentages of inhibition for each compound are displayed in .
Figure 3 Comparison of the percentages of inhibition of H37Ra growth for 4 Enamine test compounds and rifampicin (positive control) at 10 μg/mL and 0.1 μg/mL. Results are shown as averages of ±SD of 4 independent experiments.
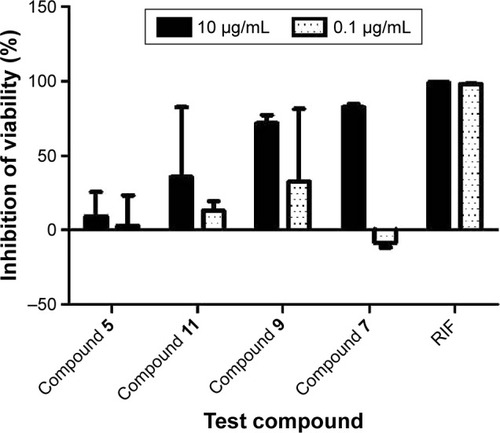
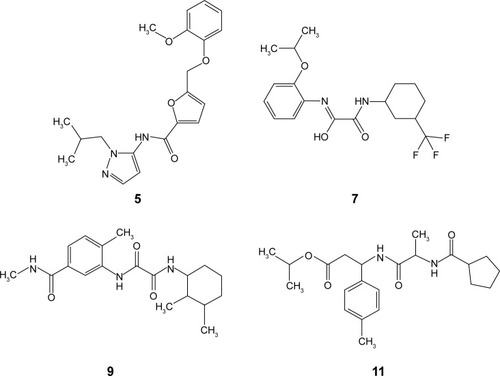
shows the percentages of H37Ra growth inhibition for 4 () of the 17 compounds tested. Although predicted to bind favorably with BioA, the remaining 13 compounds (not shown) demonstrated minuscule activities. These results demonstrate the utility of whole-cell assay in confirming the bioactivity of hit compounds obtained from structure-based methods. Possibly, the high-affinity hits were not able to penetrate the characteristic thick, waxy lipid layer of the mycobacterium. Compound 7 ((Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl) cyclohexyl)amino)acetimidic acid) was shown to be the most bioactive among all the compounds tested. Although it apparently lost its bioactivity at 0.1 μg/mL or at 0.27 μM, it exhibited the highest percentage of H37Ra growth inhibition (82.55%) at 10 μg/mL or at 26.85 μM. In subsequent experiments involving 6 fivefold dilutions starting from 125 μM to 0.04 μM, the minimum inhibitory concentration of 7 was found to be ~25 μM. The other active hits include compounds 9 (N1-(2,3-dimethylcyclohexyl)-N2-(2-methyl-5-(methylcarbamoyl)phenyl)oxalamide), 11 (isopropyl 3-(2-(cyclopentanecarboxamido)propanamido)-3-(p-tolyl) propanoate), and 5 (N-(1-isobutyl-1H-pyrazol-5-yl)-5-((2-methoxyphenoxy)methyl)furan-2-carboxamide), in order of decreasing activity.
Figure 4 Chemical structures of bioactive hits.
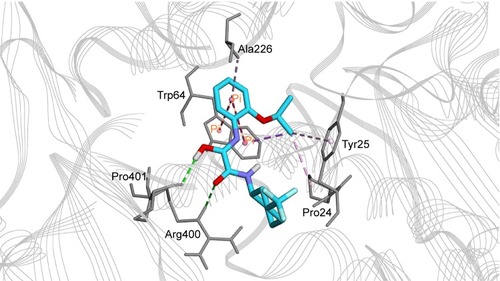
The most active hit, compound 7 ( and ), notably exhibited interaction with PLP cofactor and with prominent residues at the active site, namely Trp64, Trp65, Tyr157, Lys283, Arg400, and Phe402. Additionally, vdW interactions were observed between 7 and Trp65, Lys283, Tyr157, Phe402, and PLP cofactor. Other interactions that largely contributed to its binding affinity were the hydrogen bonding with the side chain of Tyr407 and the pi–pi bonding with Trp64. Furthermore, the polar interaction shown between compound 7 and Arg400 may represent the salt bridge that forms between ACM and Arg400.Citation20
Figure 5 Interaction diagram for BioA–compound 7 complex. Interaction diagram legends include: 1) pink circles = residues involved in hydrogen bond, charge, or polar interactions; 2) green circles = residues involved in van der Waals interactions; 3) blue circles = water molecules; 4) blue dashed arrow directed toward the electron donor = hydrogen bonding with amino acid side chains; and 5) orange line with symbols = pi interactions.
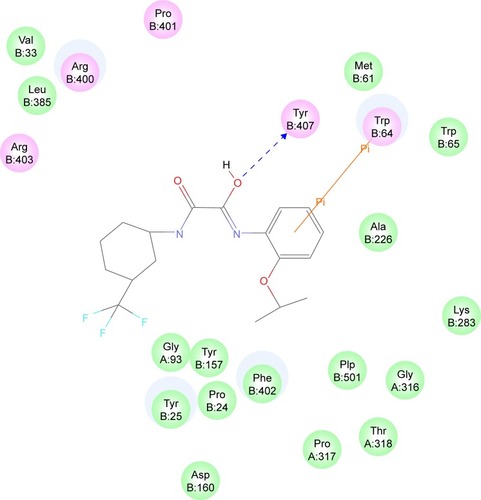
Figure 6 Binding mode of compound 7 in BioA active site. Compound 7 (cyan carbon atoms) and the key interacting residues (gray atoms) are shown in sticks. Hydrogen bonds are displayed as green dashed lines, while hydrophobic interactions are displayed as pink dashed lines, and pi-interaction pairs are connected by orange lines.
Conclusion
Computer-aided approaches to drug discovery were utilized in this study in order to discover potential novel compounds against TB. Specifically, virtual screening, molecular docking, and in silico ADMET calculations were performed on Enamine REAL database containing 4.5 million compounds. Out of 45 high-scoring, high-binding compounds with favorable in silico drug-like properties, 17 compounds were tested for their in vitro bioactivities against Mtb H37Ra using REMA. Four compounds displayed encouraging bioactivities, with compound 7 ((Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl)cyclohexyl)amino)acetimidic acid) being the most active. The docking studies showed that 7 interacts with several pertinent residues in the active site of BioA, particularly forms hydrogen bonding with the side chain of Tyr407 and pi–pi bonding with Trp64. These encouraging results should prompt follow-up lead optimization studies to further improve the potency of the identified hits.
Acknowledgments
This study was supported by the Office of the Vice President for Academic Affairs (OVPAA), University of the Philippines System under the Emerging Inter-Disciplinary Research (EIDR) program (OVPAA-EIDR 12-001-121102).
Supplementary material
Table S1 Two-dimensional structures of the inhibitor ACM, as well as the top 17 Enamine compounds generated from in silico studies
Disclosure
The authors report no conflicts of interest in this work.
References
- WorkmanPHow much gets there and what does it do?: the need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and developmentCurr Pharm Des2003911 891 90212678873
- DiMasiJAHansenRWGrabowskiHGThe price of innovation: new estimates of drug development costsJ Health Econ2003222 151 18512606142
- SongCMLimSJTongJCRecent advances in computer-aided drug designBrief Bioinform2009105 579 59119433475
- KlebeGVirtual ligand screening: strategies, perspectives and limitationsDrug Discov Today20061113–14 580 59416793526
- HouTXuXRecent development and application of virtual screening in drug discovery: an overviewCurr Pharm Des2004109 1011 103315078130
- KapetanovicIMComputer-aided drug discovery and development (CADDD): in silico-chemico-biological approachChem Biol Interact20081712 165 17617229415
- WHO [webpage on the Internet]Global tuberculosis report 2015GenevaWHO2015 Available from: http://apps.who.int/iris/bitstream/10665/191102/1/9789241565059_eng.pdf?ua=1Accessed November 6, 2015
- BillonesJBCarrilloMCOOrganoVGToward antituberculosis drugs: in silico screening of synthetic compounds against Mycobacterium tuberculosis l,d-transpeptidase 2Drug Des Dev Ther201610 1147 1157
- BillonesJBCarrilloMCOrganoVMacalinoSJYEmnacenISyJBVirtual screening against Mtuberculosis 7,8-diaminopelargonic acid synthase (MtbBioA) and in silico toxicity evaluation of top hitsCurr Enzym Inhib2014102 105 112
- BillonesJBCarrilloMCOOrganoVGMacalinoSJYEmnacenIASyJBAVirtual screening against Mycobacterium tuberculosis lipoate protein ligase B (MtbLipB) and in silico ADMET evaluation of top hitsOrient J Chem2013294 1457 1468
- UyVCCBillonesJBTowards antituberculosis drugs: virtual screening for potential inhibitors of pantothenate synthetase of Mycobacterium tuberculosisPhil Sci Lett201252 122 130
- YangCTMBillonesJBTowards antituberculosis drugs: molecular docking of curcumin and its analogues to pantothenate synthetasePhil J Sci20121412 187 196
- BillonesJBValleAMFStructure-based design of inhibitors against maltosyltransferase GlgEOrient J Chem2014303 1137 1145
- ClavioNABBillonesJBVirtual screening against Mycobacterium tuberculosis isocitrate lyase and in silico ADME-Tox evaluation of top hitsJ Chem Pharm Res2014610 727 738
- CordovaDLMAbuelRJDGalinganaMOVillanuevaLABillonesJBPiggyback drug development: molecular docking of Entacapone analogues as direct M. tuberculosis InhA inhibitorsJ Chem Pharm Res201575 636 642
- SampacoABIIIBillonesJBVirtual screening of natural products, molecular docking and dynamics simulations on M. tuberculosis S-adenosyl-L-homocysteine hydrolaseOrient J Chem2015314 1859 1865
- BillonesJBReverse docking study unravels the potential Mycobacterium tuberculosis enzyme targets of Agelasine FOrient J Chem2016322 851 858
- BhorVMDevSVasanthakumarGRSuroliaASpectral and kinetic characterization of 7,8-diaminopelargonic acid synthase from Mycobacterium tuberculosisIUBMB Life2006584 225 23316754301
- KäckHSandmarkJGibsonKSchneiderGLindqvistYCrystal structure of diaminopelargonic acid synthase: evolutionary relationships between pyridoxal-5′-phosphate-dependent enzymesJ Mol Biol19992914 857 87610452893
- DeySLaneJMLeeRERubinEJSacchettiniJCStructural characterization of the Mycobacterium tuberculosis biotin biosynthesis enzymes 7,8-diaminopelargonic acid synthase and dethiobiotin synthetaseBiochemistry20104931 6746 676020565114
- Woong ParkSKlotzscheMWilsonDJEvaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expressionPLoS Pathog201179 e100226421980288
- MannSPlouxO7,8-Diaminoperlargonic acid aminotransferase from Mycobacterium tuberculosis, a potential therapeutic target: characterization and inhibition studiesFEBS J200627320 4778 478916984394
- KeerJSmeuldersMJGrayKMWilliamsHDMutants of Mycobacterium smegmatis impaired in stationary-phase survivalMicrobiology2000146Pt 9 2209 221710974108
- RendinaARTaylorWSGibsonKThe design and synthesis of inhibitors of dethiobiotin synthetase as potential herbicidesPestic Sci1999553 236 247
- OkamiYKitaharaTHamadaMNaganawaHKondoSStudies on a new amino acid antibiotic, amiclenomycinJ Antibiot (Tokyo)1974279 656 6644436150
- SandmarkJMannSMarquetASchneiderGStructural basis for the inhibition of the biosynthesis of biotin by the antibiotic amiclenomycinJ Biol Chem200227745 43352 4335812218056
- ShiCGedersTWParkSWMechanism-based inactivation by aromatization of the transaminase BioA involved in biotin biosynthesis in Mycobacterium tuberculosisJ Am Chem Soc201113345 18194 1820121988601
- BermanHMThe Protein Data BankNucleic Acids Res2000281 235 24210592235
- Dassault Systèmes BIOVIADiscovery Studio Modeling EnvironmentSan DiegoDassault Systèmes2015
- MurrayCWReesDCThe rise of fragment-based drug discoveryNat Chem200913 187 19221378847
- PetitJMeuriceNKaiserCMaggioraGSoftening the Rule of Five – where to draw the line?Bioorg Med Chem20122018 5343 535122222160
- DaiRWilsonDJGedersTWAldrichCCFinzelBCInhibition of Mycobacterium tuberculosis transaminase BioA by aryl hydrazines and hydrazidesChembiochem2014154 575 58624482078
- PetheKSequeiraPCAgarwallaSA chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacyNat Commun20101 5720975714
- KohanskiMADwyerDJHayeteBLawrenceCACollinsJJA common mechanism of cellular death induced by bactericidal antibioticsCell20071305 797 81017803904
- PalominoJCMartinACamachoMGuerraHSwingsJPortaelsFResazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosisAntimicrob Agents Chemother2002468 2720 272212121966
- PalominoJCMartinAPortaelsFRapid drug resistance detection in Mycobacterium tuberculosis: a review of colourimetric methodsClin Microbiol Infect2007138 754 76217378933