
Abstract
The treatment of melanoma has improved markedly over the last several years with the advent of more targeted therapies. Unfortunately, complex compensation mechanisms, such as those of the mitogen-activated protein kinase (MAPK) pathway, have limited the clinical benefit of these treatments. Recently, a better understanding of melanoma resistance mechanisms has given way to intelligently designed multidrug regimes. Herein, we review the extensive pathways of BRAF inhibitor (vemurafenib and dabrafenib) resistance. We also review the advantages of dual therapy, including the addition of an MEK inhibitor (cobimetinib or trametinib), which has proven to increase progression-free survival when compared to BRAF inhibitor monotherapy. Finally, this review touches on future treatment strategies that are being developed for advanced melanoma, including the possibility of triple therapy with immune checkpoint inhibitors and the work on optimizing sequential therapy.
Introduction
Traditional chemotherapies have been well studied for metastatic melanoma with no evidence supporting survival benefit. Melanoma oncogenesis involves both DNA damage from ultraviolet light and genetic predispositions.Citation1,Citation2 In the last several years, successful targeted therapies have revolutionized the treatment of advanced melanomas by targeting the mitogen-activated protein kinase (MAPK) pathway. The MAPK pathway is a critical regulator of cell proliferation and survival. BRAF and its downstream target, MEK, are kinases in the MAPK pathway, and therefore play an important role in cell proliferation.Citation3,Citation4 The discovery of somatic BRAF V600 mutations in melanomas initiated the development of targeted therapies. BRAF inhibitors were the first pharmacological agents used clinically, but tumor resistance has limited their benefit. To overcome these resistance mechanisms, MEK inhibitors have been used in combination and their results are promising. This review explores the resistance pathways of BRAF inhibitors and the role of MEK inhibitors in combating BRAF inhibitor resistance in advanced BRAF-mutant melanomas.
The start of targeted therapy
After the BRAF V600 mutation was identified in melanoma,Citation4 BRAF inhibitors were developed for advanced melanomas harboring this mutation. In 2011, the US Food and Drug Administration (FDA) approved the BRAF inhibitor vemurafenib for treatment of unresectable or metastatic melanoma with BRAF(V600E) mutations.Citation5 In patients with advanced melanoma, the median progression-free survival (PFS) with single-agent vemurafenib ranges from 5 to 7 months, and the median overall survival is approximately 16 months, which is 7 months more than with chemotherapy.Citation6–Citation9
In 2013, dabrafenib became the second FDA-approved BRAF inhibitor with similar indications.Citation10 Although a significant difference in overall survival was not observed, patients treated with single-agent dabrafenib demonstrated an improved median PFS compared to those treated with dacarbazine.Citation11
Development of BRAF inhibitor resistance through reactivation of the MAPK pathway
Unfortunately, the clinical benefit of a BRAF inhibitor is limited by intrinsic and acquired resistance. Reactivation of the MAPK pathway is a major contributor to treatment failure in BRAF-mutant melanoma (). In fact, a study of resistance mechanisms showed that reactivation of MAPK signaling drives BRAF inhibitor resistance in roughly 80% of melanoma tumors.Citation12,Citation13
Figure 1 Schematic diagram representing MEK inhibitor-sensitive reactivation of MAPK signaling following BRAF inhibitor resistance.
Abbreviations: NF1, neurofibromin-1; RTK, receptor tyrosine kinase; MLK, mixed lineage kinases; ERK, extracellular-signal-regulated kinase; MAPK, mitogen-activated protein kinase.
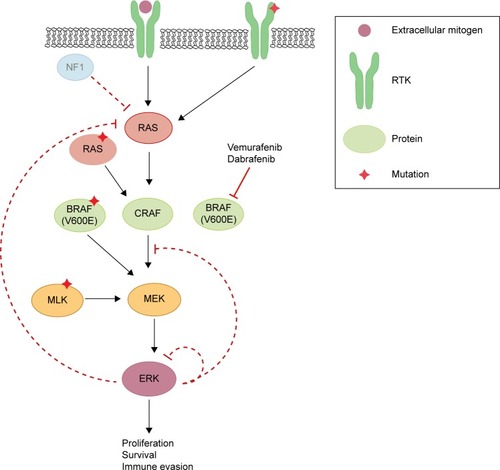
Reactivation of MAPK through receptor tyrosine kinase activation and ERK rebound
Receptor tyrosine kinases (RTKs), upstream of RAS in the MAPK pathway, consist of growth factor receptors for ligands such as epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF), and platelet-derived growth factor receptor (PDGFR). Stimulation of RTKs activates RAS, which triggers downstream signaling cascades. Mutations in the genetic coding or regulation of expression of these enzymes have been shown to induce and promote resistance to BRAF inhibition, including invasion and metastasis.Citation14–Citation17 One study demonstrated that increased levels of basic FGF induced higher FGF receptor 3 (FGFR3) activity, thus reactivating MAPK in vemurafenib-resistant cells in culture.Citation17 The authors also discovered a constitutively active mutant of FGFR3 promoted BRAF inhibitor resistance through the MAPK pathway. Adding bFGF led to RAS activation, upregulated extracellular-signal-regulated kinase (ERK) activation, and was responsive to both pan-RAF and MEK inhibitors.
Negative feedback of MAPK is directly mediated by ERK-dependent phosphorylation of enzymes in the pathway including RAF, RTK, and SOS.Citation18,Citation19 Additionally, ERK activation induces expression of negative regulators of MAPK.Citation20,Citation21 In BRAF(V600E) cancers, the MAPK pathway is hyper-stimulated, which suppresses ERK-dependent feedback inhibition. When a BRAF(V600E)-mutant melanoma is treated with a RAF inhibitor, there is induction of RAS-GTP accompanied by a rebound in phospho-ERK (pERK).Citation22 This rise in RAS-GTP levels promotes CRAF dimerization and the subsequent phosphorylation of MEK, and thereby decreases the effectiveness of RAF inhibitors. Although CRAF dimers are insensitive to vemurafenib, ERK rebound through CRAF remains sensitive to MEK inhibition.
Further findings demonstrate that various growth factor ligands (EGF, hepatocyte growth factor [HGF], neuregulin [NRG], FGF) can antagonize vemurafenib sensitivity via ligand-induced sensitization,Citation22,Citation23 which promotes ERK rebound and desensitization to BRAF inhibition.Citation24 These findings suggest that enhancement of RTK signaling is due to relief of ERK-dependent feedback inhibition. Combined inhibition of the MAPK pathway could theoretically increase the degree of response duration compared to RAF inhibition alone.
Reactivation of MAPK through RAS
HRAS, KRAS, and NRAS are RAS isoforms within the MAPK pathway. While BRAF is mutated in 50% of melanomas, NRAS and KRAS are mutated in about 20% and 2%, respectively.Citation25 Although BRAF inhibitors can induce metastasis in RAS mutants by reactivation of MEK/ERK signaling, this mechanism can be blocked by MEK inhibition.Citation25 Mutations in KRAS and NRAS reactivate the MAPK pathway, activating tumor growth as well as accelerating preexisting but previously benign secondary tumors.Citation26,Citation27 Melanomas were found to acquire resistance to BRAF(V600E) inhibition by acquisition of activating NRAS mutations as well as NRAS upregulation through increased copy number.Citation16 Additionally, although NRAS mutations concurrent with BRAF V600 mutations are rare, they can promote BRAF inhibitor resistance through reactivation of MAPK via preference for CRAF.Citation28 Others have shown that some mutations in BRAF or KRAS alone do not induce melanoma, but dual mutations can promote CRAF-dependent resistance.Citation29 Knockdown of NRAS in mutant melanoma cells resulted in decreased pERK with a predominantly apoptotic response, suggesting MAPK dependence in mutated NRAS melanomas.Citation30 Indeed, sensitization to MEK inhibition is a hallmark trait of NRAS-mutated BRAF inhibitor-resistant cells.Citation16
Reactivation of MAPK through silencing of NF1
RAS proteins are activated when guanine exchange factors bind GTP and inactivated by GTPase-activating proteins. Active RAS facilitates dimerization and activation of RAF kinases, which stimulates MEK and ERK. RAS proteins are inactivated by GTPase-activating proteins such as neurofibromin-1 (NF1), a GTP-binding factor that facilitates GTP hydrolysis.Citation31 Consequently, NF1 is a negative regulator of the MAPK pathway. Genome-scale RNA interference and screening of melanoma tumors identified inactivation of NF1 as a mechanism of resistance to BRAF inhibition,Citation32 due to hyperactivation through CRAF dimerization.Citation32–Citation34 Furthermore, mutant NF1 alleles identified in melanomas with relatively short response times were thought to confer either a selective intrinsic or acquired advantage. NF1 silencing in BRAF-mutant melanoma cell lines in the presence of a BRAF inhibitor led to increased active GTP-bound RAS, sustained phosphorylation of ERK, and proliferation.Citation34 Mutants with complete loss of NF1 exhibited resistance to BRAF inhibitors. However, NF1 null cells treated with a MEK inhibitor displayed decreased proliferation, highlighting MEK dependence.Citation34
Reactivation of MAPK through RAF
Mutations inducing upregulation of BRAF(V600E) were also found to confer resistance to BRAF inhibitor therapy. A copy-number gain of mutated BRAF(V600E) provided a form of resistance yet was still dependent on MEK/ERK signaling.Citation35 Splice variants of BRAF(V600E) were found to be a driving force in vemurafenib resistance in mutant melanoma cell culture as well as in patient tumors.Citation36 The causative alternative splicing event was shown to be an in-frame deletion confined to the mutant BRAF allele. Structurally, this promoted constitutive dimerization of mutant BRAF which bestowed BRAF inhibitor resistance, even in the absence of RAS activation. This mutant also retained sensitivity to MEK inhibition, demonstrating that additional downstream pathway inhibitors are necessary to delay or inhibit resistance. Furthermore, potential resistance to BRAF inhibition could be induced by switching tumor dependency from BRAF to increasing expression of CRAF.Citation37
Paradoxical oncogenesis is a concern in BRAF inhibitor-treated melanoma due to transactivation of RAF isoforms. Vemurafenib and dabrafenib bind to the highly active monomeric mutant BRAF(V600) to inhibit its function. However inhibitor-bound mutant BRAF can still dimerize with uninhibited CRAF to activate MAPK, consequently leading to proliferation and tumor progression,Citation38,Citation39 highlighting the importance of additional downstream inhibitors.
Reactivation of MAPK through MEK and MEK-activating kinase
Mixed lineage kinases (MLK1–4) are MEK kinases that are able to independently reactivate the MEK/ERK pathway, even in the presence of RAF inhibitors. Studies of acquired resistance in melanoma tumors demonstrated that BRAF inhibitor resistance correlates with MLK upregulation,Citation40,Citation41 as well as a gain of function mutation in MLK1,Citation41 leading to patient tumor progression. Furthermore, MLK expression in the presence of vemurafenib prevented apoptosis, contributing an additional mechanism of survival advantage by MLK in BRAF inhibitor resistance.Citation41
Immune evasion and survival through alteration of tumor microenvironment
Recent data suggest that BRAF(V600E) contributes to immune escape.Citation42,Citation43 Expression of melanoma antigens Mart1, Tyrp1, Tyrp2, and GP100 were significantly increased after treatment of BRAF-mutant melanoma cell lines with BRAF inhibitor, facilitating T-cell cytotoxicity.Citation42 BRAF inhibition was associated with a decrease in immunosuppressive cytokines,Citation42,Citation43 as well as a significant increase in CD8+ T-cell infiltrate,Citation42,Citation44 and T-cell recognition of melanoma.Citation45 Thus, compared to no treatment, BRAF inhibitor therapy provides a more favorable tumor microenvironment, supporting the idea of potential synergy of BRAF-targeted therapy and immunotherapy.Citation46
PDL1 is an inducible cell surface protein, one of two ligands for programmed-death receptor 1 (PD1), a molecule that negatively regulates T-cell immune responses. Increased PDL1 expression in cancer cells is a significant escape mechanism from host immunity.Citation47 Although BRAF inhibition induced immune evasion through increased PDL1, BRAF inhibitor-treated tumors exhibited increased markers for T-cell exhaustion signifying a dysfunctional immune response.Citation42 At time of resistance, melanoma tumor antigen expression and CD8+ T-cell infiltrate are decreased. As MAPK reactivation leads to higher PDL1 expression in BRAF inhibitor-resistant cells, MEK inhibition expectedly and significantly reduced PDL1 in BRAF inhibitor-resistant melanoma cellsCitation48,Citation49 and induced apoptosis.Citation48 In another cancer model, MEK inhibition reversed PDL1-mediated inhibition of cytotoxic T-cell function against acute myeloid leukemia cells.Citation50
Use of MEK inhibitors in BRAF-mutant melanoma
Given that the majority of BRAF inhibitor resistance occurs through reactivation of MAPK, several potent, non-ATP competitive MEK inhibitors were developed (eg, trametinib and cobimetinib)Citation51 that show no off-target effects on other kinases and are currently being used in clinical trials. MEK inhibitors are able to target MAPK-dependent tumors and exhibit distinct efficacies against BRAF- and KRAS-mutant melanomas.Citation52,Citation53
Cobimetinib is a potent selective oral MEK inhibitor with antitumor activity and it demonstrates 100-fold stronger potency against mutated BRAF-mutant versus KRAS-mutant cell lines.Citation52 Cobimetinib binds strongly to pMEK, making it highly potent in inhibiting BRAF(V600E) melanoma, since mutant BRAF constantly produces high levels of pMEK. On the other hand, trametinib displays higher binding affinity to unphosphorylated MEK.Citation54 However, both inhibitors are able to inhibit downstream ERK signaling, particularly effective in BRAF-mutant tumor cells. Studies reveal that BRAF inhibitor-resistant tumor cells are highly sensitive to MEK inhibition and demonstrate that targeted pharmacological MEK inhibition may be a highly effective therapeutic alternative in BRAF inhibitor-resistant melanoma.Citation53 In light of these findings, focus has turned to dual inhibition of BRAF and its downstream target, MEK.
Clinical trials
Single-agent MEK inhibitors
Trametinib, a competitive allosteric MEK inhibitor, demonstrated promise as monotherapy in a Phase III trial. The median PFS of patients who received trametinib was significantly longer than that of patients who received chemotherapy (4.8 vs 1.5 months; hazard ratio [HR] 0.45; P<0.001). At 6 months, the rate of overall survival was significantly higher as well in the trametinib group (81% vs 67%; HR 0.54; P=0.01).Citation55 In a small sample cohort, selu-metinib, another MEK inhibitor, elicited tumor suppression in BRAF-mutated melanomas, but only in low phosphor-Akt melanomas. This observation further supports that resistance can also be caused by overactivation of the PI3K/Akt pathway.Citation56 Although cobimetinib has not been evaluated as a single agent in clinical trials, the maximum tolerated dose is 60 mg daily on the 21-day on/7-day off dosing schedule. At this dose, cobimetinib is generally well tolerated and the most frequent adverse effects include diarrhea, rash, nausea, fatigue, dry skin, and peripheral edema.Citation57
Combination therapy
The results of combination therapy of MEK inhibitors and BRAF inhibitors are promising. New clinical trials confirm improved PFS with concurrent inhibition of BRAF and MEK. coBRIM (NCT01689519), a Phase III, randomized study, compared patients with BRAF-mutated melanomas treated with vemurafenib 960 mg twice daily versus vemu-rafenib plus cobimetinib 60 mg daily for 21 days followed by 7 days off. The group on combination therapy displayed prolonged median PFS (9.9 months vs 6.2 months). At the time of interim analysis, the median overall survival had yet to be reached for both groups ().Citation58 The coBRIM study is scheduled to conclude in 2017 and will provide highly anticipated data on the survival benefits of combination vemurafenib and cobimetinib therapy. With the promising results of the coBRIM study, the FDA in November 2015 approved cobimetinib for its utility in combination with vemurafenib in patients with advanced BRAF V600 mutation-positive melanomas.
Table 1 Summary of the clinical trials and outcome measures for combination vemurafenib and cobimetinib therapy
Other clinical trials focused on different BRAF and MEK inhibitor combinations suggested similar findings.Citation59–Citation61 In fact, the latest Phase III clinical trial of dabrafenib (BRAF inhibitor) and trametinib versus monotherapy with vemurafenib showed increased median PFS (11.4 months vs 7.3 months; HR 0.56; 95% confidence interval [CI]: 0.46–0.69; P<0.001) and improved overall survival at 12 months with combination therapy (72% vs 65%; HR 0.69; 95% CI: 0.53–0.89; P=0.005).Citation60 Additionally, the double-blinded COMBI-d trial compared dabrafenib and trametinib vs dabrafenib and placebo. Interim results demonstrated an increased median PFS as well (9.3 months vs 8.8 months; HR 0.75; 95% CI: 0.57–0.99; P=0.035).Citation62
Cutaneous adverse events
An additional benefit of combination therapy is the decreased frequency of cutaneous adverse events compared to single-agent MEK or BRAF inhibitor therapy.Citation63 Although BRAF inhibitors decrease MAPK pathway activity in BRAF-mutant cells, paradoxical activation can take place in BRAF wild-type cells with upstream mutations. This leads to the development of keratoacanthomas and squamous cell carcinomas early in BRAF inhibitor treatment.Citation27,Citation64,Citation65 Co-targeting with MEK inhibitors appears to decrease this undesired proneoplastic adverse effect that BRAF inhibitors have on keratinocytes.Citation26 For example, only 3.5% of patients on combination therapy developed cutaneous squamous cell carcinoma and keratoacanthoma, whereas 19.7% of those on vemurafenib developed these adverse events.Citation58 Therefore, transitioning from monotherapy to combination therapy would decrease concerns for undesired cutaneous adverse events.
Future studies
With recent positive outcomes, dual BRAF and MEK inhibition therapy will likely be the future of advanced melanoma therapy. Further clinical trials are under way and we anticipate that combination therapy will become the standard of care over monotherapy.
Specific patient populations
Investigation into the potential of dual BRAF and MEK inhibitor therapy in specific patient populations is currently under way. coBRIM-B, a multicenter Phase II study, plans to evaluate the utility of combination vemurafenib and cobimetinib in metastatic melanoma patients with brain involvement (NCT02230306). Additionally COMBI-AD, a double-blinded Phase II study, is currently evaluating the efficacy of combination dabrafenib and trametinib as adju-vant treatment in patients with completely resected advanced melanoma (NCT01682083). These studies may broaden the utility of combination therapy in melanoma patients.
Sequential therapy
The future of melanoma therapy may not hinge solely on combination therapy, but also on finding an optimal sequential regime to combat melanoma as it evolves.Citation66 In a Phase Ib study of 66 patients with advanced melanoma who progressed on vemurafenib, transitioning to combination therapy resulted in a limited response with a median PFS of 2.8 months. The clinical significance of this modest response is unclear since the study was not designed to directly compare effects of sequential therapy with a combination regime. However, the response is undeniably worse than the response seen with combination vemurafenib and cobime-tinib in BRAF inhibitor-naïve patients, who had a median PFS of 13.7 months.Citation67
Another study looked at the effect of trametinib in patients who were previously treated with BRAF inhibitor vs those who were treated with chemotherapy and/or immunotherapy. Trametinib monotherapy had minimal clinical activity when administered as sequential therapy in patients with BRAF-mutant melanoma who had failed BRAF inhibitor therapy.Citation68 Therefore, the therapeutic role of adding a MEK inhibitor after BRAF inhibitor resistance has already occurred is unclear, and further investigation on sequential therapy is required to address this option fully.
Paradox-breakers
Recently, new compounds have been developed that inhibit both BRAF and CRAF as well as SRC family kinases (SFKs) termed “paradox-breakers”. These inhibitors were shown to be active in treatment-naïve BRAF-mutant tumors, BRAFi-resistant tumors, and cells isolated from a patient with BRAF/MEK inhibitor-resistant melanoma.Citation69,Citation70 The MAPK pathway was upregulated in each of these cases, demonstrating that these inhibitors are useful in the context of high pERK levels. Moreover, NRAS-mutant tumors reliant on CRAF are highly sensitive to these inhibitors. Future clinical studies will be required to ascertain treatment strategies for these compounds.
Resistance mechanisms not responsive to BRAF or MEK inhibitors
Despite these clinical benefits, the emergence of resistance to BRAF inhibitors and eventually to MEK inhibitors restricts the therapeutic efficacy of these kinase inhibitors (). In an attempt to overcome this resistance, more potential molecules targeting other aberrant signaling pathways are in clinical development.Citation71,Citation72
Figure 2 Schematic diagram representing aberrant signaling pathways responsible for resistance to BRAF or MEK inhibitors in metastatic melanoma and pharmacological strategies to overcome this resistance.
Abbreviations: PDGFR, platelet-derived growth factor receptor; IGF, insulin-like growth factor; ERK, extracellular-signal-regulated kinase.
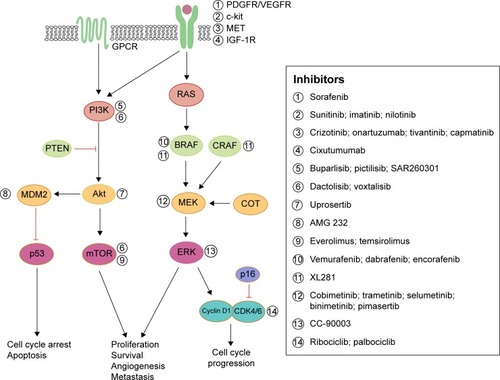
A myriad of studies have determined pathways other than MAPK that can confer resistance. Those upregulated mechanisms are briefly discussed here. An increase in RTK ligands, either through autocrine production from tumor cells, paracrine secretion from stroma, or systemic production, promotes resistance to kinase inhibitors.Citation24,Citation73 BRAF inhibitor resistance mediated by EGFR was shown in both cell culture studies and patient melanoma.Citation15,Citation74 Similarly, IGF-1RCitation75 and MET receptorCitation23 were found to reactivate both the MAPK and PI3K/AKT pathway, suggesting that inhibition of MEK is only one step of overcoming resistance to BRAF inhibition. Indeed, patient-derived samples revealed tumor-associated upregulation of PDGFR3Citation16,Citation74 in vemurafenib-resistant cells, which was shown to be SOX10 and TGFBR2 dependent.Citation74 MEK inhibition is ineffective in these resistant strains, demonstrating that MEK/MAPK is not the key downstream pathway here. Both cyclin D1 as well as PTEN were implicated in BRAF inhibitor resistance.Citation76,Citation77
Single codon mutations in MEK were found in tumors from relapsed patients while on treatment with a BRAFCitation28,Citation78 or MEK inhibitor.Citation79 A recent study of vemurafenib-resistance tumors revealed a MEK mutation at codon 56 which produced the highest levels of pERK (compared to other MEK mutations).Citation28 Overall, these MEK mutations confer BRAF inhibitor resistance and exhibited robust resistance to MEK inhibition,Citation40,Citation79 highlighting the necessity for further downstream inhibition at the level of ERK.Citation80 As others have suggested,Citation16 these findings support specific treatment strategies for patients who relapse on vemurafenib and require precise combinations of targeting agents to optimize inhibition of proliferation.
With these sources of dual-therapy resistance, targeting BRAF and/or MEK may also require targeting other upregulated enzymes to achieve durable therapeutic responses.Citation81 Clinical trials are currently under way to assess safety and efficacy of MEK inhibition (cobimetinib, pimasertib, trametinib) combined with single-agent PI3K/Akt/mTOR pathway inhibitors (BKM120, GDC-0941, uprosertib, BEZ235, SAR245409) (NCT01363232, NCT01138085, NCT01337765, NCT01390818, and NCT00996892, respectively). Further, trials simultaneously inhibiting both BRAF and MEK, as well as a third inhibitor targeting individual enzymes (Akt, MDM2, Bcl-2, or MET) are also taking place for advanced melanoma patients (NCT01902173, NCT02110355, NCT01989585, or NCT01974258, respectively). Finally, a Phase I trial is under way in relapsed or refractory BRAF(V600)-mutated melanoma with a selective ERK inhibitor, CC-90003 (NCT02313012).
In a different combined-therapy approach using a preclinical model, intermittent combination therapy with MEK inhibitor (cobimetinib) and PI3K inhibitor (GDC-0941) was sufficient to demonstrate a robust antitumor effect against BRAF/KRAS-mutated melanoma, compared to continuous exposure of these agents.Citation82 Taking one step further, “adaptive sequential therapy”Citation83 is a new therapeutic strategy of using targeted inhibitors based upon the patients’ resistance mechanism. An ongoing Phase II clinical trial for advanced metastatic melanoma provides BRAF inhibitor therapy until progression; upon progression, patients will be treated with an interventional drug targeting enzymes within the upregulated resistance pathway (NCT01820364).
Targeted treatment in combination with immunotherapy
In spite of the impressive results of targeted inhibition of MAPK, further advancements are needed to improve treatment outcomes for all patients in metastatic melanoma. As immunotherapy has displayed positive results in melanoma,Citation46,Citation84,Citation85 it was suggested to synergistically augment the immune system by combining immune checkpoint blockades with MAPK inhibition.Citation86,Citation87 A representative combination therapy is the use of BRAF inhibitor combined with antibodies that prevent inhibitory signaling, such as anti-CTLA-4 (ipilimumab) or anti-PD1 (pembrolizumab). The first clinical trial combining MAPK inhibition with immunotherapy involved ipilimumab and vemurafenib (NCT01400451); however, this combination therapy was terminated due to substantial liver toxicities.Citation88 Investigations with other combinations of MAPK inhibitors, anti-PDL1 immunotherapies, cytokines (IL-2, IFN-alpha 2b), or adoptive cell transfer therapy are still ongoing in clinical trials.Citation89
Although immunotherapies are highlighted by recent success in advanced melanoma,Citation89–Citation92 toxic effects in combination with kinase-targeted therapy remain unpredictable. Thus, the dosing, timing, and adverse effects of combination treatments involving immunotherapy need to be carefully studied. Through further studies, prolonged antitumor response using combination MAPK inhibition with immunomodulators may be achievable for patients with advanced melanoma. With their high antitumor activity and synergistic properties, these therapies have a high potential for improving clinical outcomes.
Conclusion
With the discovery of BRAF-mutant melanomas, the development of new targeted therapeutics has increased tremendously in the last several years. These drugs hold great potential in improving the prognosis of advanced melanoma, but are limited by intrinsic and acquired resistance. BRAF inhibitors have demonstrated tumor response and increased median PFS in advanced BRAF-mutant melanomas. However, their utility is limited by numerous, complex, and often overlapping resistance pathways. Usage of MEK inhibitors in conjunction with BRAF inhibitors offers a method of combating this resistance. However, acquired resistance may also hinder dual therapy. Whether the best strategy to prolong survival is through optimization of triple therapy or sequential therapy will need to be further explored.
Disclosure
The authors report no conflicts of interest in this work.
References
- JhappanCNoonanFPMerlinoGUltraviolet radiation and cutaneous malignant melanomaOncogene200322203099311212789287
- MaverakisEMiyamuraYBowenMPCorreaGOnoYGoodarziHLight, including ultravioletJ Autoimmun2010343J247J25720018479
- PeyssonnauxCEychèneAThe Raf/MEK/ERK pathway: new concepts of activationBiol Cell2001931–2536211730323
- DaviesHBignellGRCoxCMutations of the BRAF gene in human cancerNature2002417689294995412068308
- KimGMcKeeAENingYMFDA Approval summary: vemu-rafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutationClin Cancer Res201420194994500025096067
- ChapmanPBHauschildARobertCBRIM-3 Study GroupImproved Survival with vemurafenib in melanoma with BRAF V600E mutationN Engl J Med2011364262507251621639808
- SosmanJAKimKBSchuchterLSurvival in BRAF V600-mutant advanced melanoma treated with vemurafenibN Engl J Med2012366870771422356324
- McArthurGAChapmanPBRobertCSafety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label studyLancet Oncol201415332333224508103
- RobertCThomasLBondarenkoIIpilimumab plus dacarba-zine for previously untreated metastatic melanomaN Engl J Med2011364262517252621639810
- FDA Approval for Dabrafenib [webpage on the Internet]BethesdaNational Cancer Institute [updated January 16, 2014]. Available from: http://www.cancer.gov/about-cancer/treatment/drugs/fda-dabrafenibAccessed October 4, 2015
- HauschildAGrobJJDemidovLVDabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trialLancet2012380983935836522735384
- RizosHMenziesAMPupoGMBRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impactClin Cancer Res20142071965197724463458
- SullivanRJFlahertyKMAP kinase signaling and inhibition in melanomaOncogene201332192373237922945644
- AbelEVBasileKJKugelCH3rdMelanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3J Clin Invest201312352155216823543055
- GirottiMRPedersenMSanchez-LaordenBInhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanomaCancer Discov20133215816723242808
- NazarianRShiHWangQMelanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulationNature2010468732697397721107323
- YadavVZhangXLiuJReactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanomaJ Biol Chem201228733280872809822730329
- DoughertyMKMüllerJRittDARegulation of Raf-1 by direct feedback phosphorylationMol Cell200517221522415664191
- DouvilleEDownwardJEGF induced SOS phosphorylation in PC12 cells involves P90 RSK-2Oncogene19971543733839242373
- EblaghieMCLunnJSDickinsonRJNegative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryosCurr Biol200313121009101812814546
- HanafusaHToriiSYasunagaTNishidaESprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathwayNat Cell Biol200241185085812402043
- LitoPPratilasCAJosephEWRelief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomasCancer Cell201222566868223153539
- StraussmanRMorikawaTSheeKTumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretionNature2012487740850050422763439
- ParaisoKHFedorenkoIVCantiniLPRecovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapyBr J Cancer2010102121724173020531415
- Sanchez-LaordenBVirosAGirottiMRBRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signalingSci Signal20147318ra3024667377
- OberholzerPAKeeDDziunyczPRAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitorsJ Clin Oncol201230331632122067401
- SuFVirosAMilagreCRAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitorsN Engl J Med2012366320721522256804
- TrunzerKPavlickACSchuchterLPharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanomaJ Clin Oncol201331141767177423569304
- HeidornSJMilagreCWhittakerSKinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAFCell2010140220922120141835
- EskandarpourMKiaiiSZhuCCastroJSakkoAJHanssonJSuppression of oncogenic NRAS by RNA interference induces apoptosis of human melanoma cellsInt J Cancer20051151657315688405
- LiYBollagGClarkRSomatic mutations in the neurofibroma-tosis 1 gene in human tumorsCell19926922752811568247
- WhittakerSRTheurillatJPVan AllenEA genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibitionCancer Discov20133335036223288408
- WanPTGarnettMJRoeSMMechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAFCell2004116685586715035987
- NissanMHPratilasCAJonesAMLoss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependenceCancer Res20147482340235024576830
- ShiHMoriceauGKongXMelanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistanceNat Commun2012372422395615
- PoulikakosPIPersaudYJanakiramanMRAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E)Nature2011480737738739022113612
- MontagutCSharmaSVShiodaTElevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanomaCancer Res200868124853486118559533
- GibneyGTMessinaJLFedorenkoIVSondakVKSmalleyKSParadoxical oncogenesis – the long-term effects of BRAF inhibition in melanomaNat Rev Clin Oncol201310739039923712190
- FreemanAKRittAAMorrisonDKEffects of Raf dimerization and its inhibition on normal and disease-associated Raf signalingMol Cell201349475175823352452
- JohannessenCMBoehmJSKimSYCOT drives resistance to RAF inhibition through MAP kinase pathway reactivationNature2010468732696897221107320
- MarusiakAAEdwardsZCHugoWMixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitorsNat Commun20145390124849047
- FrederickDTPirisACogdillAPBRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanomaClin Cancer Res20131951225123123307859
- KhaliliJSLiuSRodríguez-CruzTGOncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanomaClin Cancer Res201218195329534022850568
- WilmottJSLongGVHowleJRSelective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanomaClin Cancer Res20121851386139422156613
- BoniACogdillAPDangPSelective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte functionCancer Res201070135213521920551059
- HodiFSO’DaySJMcDermottDFImproved survival with ipilimumab in patients with metastatic melanomaN Engl J Med2010363871172320525992
- FreemanGJLongAJIwaiYEngagement of the PD-1 immunoin-hibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activationJ Exp Med200019271027103411015443
- JiangXZhouJGiobbie-HurderAWargoJHodiFSThe activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibitionClin Cancer Res201319359860923095323
- AtefiMAvramisELassenAEffects of MAPK and PI3K pathways on PD-L1 expression in melanomaClin Cancer Res201420133446345724812408
- BerthonCDrissVLiuJIn acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitorsCancer Immunol Immunother201059121839184920814675
- RiceKDAayNAnandNKNovel carboxamide-based allosteric MEK inhibitors: discovery and optimization efforts toward XL518 (GDC-0973)ACS Med Chem Lett20123541642124900486
- HatzivassiliouGHalingJRChenHMechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancersNature2013501746623223623934108
- SolitDBGarrawayLAPratilasCABRAF mutation predicts sensitivity to MEK inhibitionNature2006439707435836216273091
- GilmartinAGBleamMRGroyAGSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacoki-netic properties for sustained in vivo pathway inhibitionClin Cancer Res2011175989100021245089
- FlahertyKTRobertCHerseyPETRIC Study GroupImproved survival with MEK inhibition in BRAF-mutated melanomaN Engl J Med2012367210711422663011
- CatalanottiFSolitDBPulitzerMPPhase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanomaClin Cancer Res20131982257226423444215
- RosenLLoRussoPMaWWA first-in-human phase 1 study to evaluate the MEK1/2 inhibitor GDC-0973 administered daily in patients with advanced solid tumors [abstract]. In: Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research; 2011 April 2–6; Orlando, FLPhiladelphiaAACRCancer Res2011718 Suppl Abstract 4716
- LarkinJAsciertoPADrénoBCombined vemurafenib and cobimetinib in BRAF-mutated melanomaN Engl J Med2014371201867187625265494
- LongGVStroyakovskiyDGogasHCombined BRAF and MEK inhibition versus BRAF inhibition alone in melanomaN Engl J Med2014371201877188825265492
- RobertCKaraszewskaBSchachterJImproved overall survival in melanoma with combined dabrafenib and trametinibN Engl J Med20153721303925399551
- FlahertyKTInfanteJRDaudACombined BRAF and MEK inhibition in melanoma with BRAF V600 mutationsN Engl J Med2012367181694170323020132
- LongGVStroyakovskyDLGogasHCOMBI-d: a randomized, double-blinded, Phase III study comparing the combination of dab-rafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients (pts) with unresectable or metastatic BRAFV600E/K mutation-positive cutaneous melanomaJ Clin Oncol2014325s suppl; abstr 9011
- SanlorenzoMChoudhryAVujicIComparative profile of cutaneous adverse events: BRAF/MEK inhibitor combination therapy versus BRAF monotherapy in melanomaJ Am Acad Dermatol201471611021109.e125440439
- HatzivassiliouGSongKYenIRAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growthNature2010464728743143520130576
- PoulikakosPIZhangCBollagGShokatKMRosenNRAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAFNature2010464728742743020179705
- CurtiBDRapid evolution of combination therapy in melanomaN Engl J Med2014371201929193025390744
- RibasAGonzalezRPavlickACombination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b studyLancet Oncol201415995496525037139
- KimKBKeffordRPavlickACPhase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitorJ Clin Oncol201331448248923248257
- GirottiMRLopesFPreeceNParadox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanomaCancer Cell2015271859625500121
- LeKBlomainESRodeckUAplinAESelective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cellsPigment Cell Melanoma Res201326450951723490205
- HaoMSongFDuXAdvances in targeted therapy for unre-sectable melanoma: new drugs and combinationsCancer Lett201535911825578781
- NiezgodaANiezgodaPCzajkowskiRNovel approaches to treatment of advanced melanoma: a review on targeted therapy and immuno-therapyBiomed Res Int2015201585138726171394
- ChengHTeraiMKageyamaKParacrine effect of NRG1 and HGF drives resistance to MEK inhibitors in metastatic uveal melanomaCancer Res201575132737274825952648
- SunCWangLHuangSReversible and adaptive resistance to BRAF(V600E) inhibition in melanomaNature2014508749411812224670642
- VillanuevaJVulturALeeJTAcquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3KCancer Cell201018668369521156289
- NathansonKLMartinAMWubbenhorstBTumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436)Clin Cancer Res201319174868487823833299
- SmalleyKSLioniMDalla PalmaMIncreased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomasMol Cancer Ther2008792876288318790768
- WagleNEmeryCBergerMFDissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profilingJ Clin Oncol201129223085309621383288
- EmeryCMVijayendranKGZipserMCMEK1 mutations confer resistance to MEK and B-RAF inhibitionProc Natl Acad Sci U S A200910648204112041619915144
- HatzivassiliouGLiuBO’BrienCERK inhibition overcomes acquired resistance to MEK inhibitorsMol Cancer Ther20121151143115422402123
- JaiswalBSJanakiramanVKljavinNMCombined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumorsPLoS One200945e571719492075
- HoeflichKPMerchantMOrrCIntermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibitionCancer Res201272121021922084396
- SpagnoloFGhiorzoPOrgianoLBRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategiesOnco Targets Ther2015815716825653539
- RosenbergSARestifoNPYangJCMorganRADudleyMEAdoptive cell transfer: a clinical path to effective cancer immunotherapyNat Rev Cancer20088429930818354418
- JohnsonLAMorganRADudleyMEGene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigenBlood2009114353554619451549
- Hu-LieskovanSRobertLHomet MorenoBRibasACombining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challengesJ Clin Oncol201432212248225424958825
- MenziesAMLongGVSystemic treatment for BRAF-mutant melanoma: where do we go next?Lancet Oncol2014159e371e38125079100
- RibasAHodiFSCallahanMKontoCWolchokJHepatotoxicity with combination of vemurafenib and ipilimumabN Engl J Med2013368141365136623550685
- McDermottDLebbéCHodiFSDurable benefit and the potential for long-term survival with immunotherapy in advanced melanomaCancer Treat Rev20144091056106425060490
- PorterDLLevineBLKalosMBaggAJuneCHChimeric antigen receptor-modified T cells in chronic lymphoid leukemiaN Engl J Med2011365872573321830940
- MaverakisECorneliusLABowenGMMetastatic melanoma – a review of current and future treatment optionsActa Derm Venereol201595551652425520039
- ShiVYTranKPatelF100% complete response rate in patients with cutaneous metastatic melanoma treated with intralesional interleukin (IL)-2, imiquimod, and topical retinoid cominbation therapy: results of a case seriesJ Am Acad Dermatol201573464565426259990