
Abstract
Wernicke encephalopathy (WE) is a life-threatening but reversible syndrome resulting from acute thiamine deficiency that is frequently overlooked and underdiagnosed. It is classically characterized by a triad of ocular dysfunction, ataxia, and altered mental status. However, less than 1/3 patients have the complete triad, so it is crucial to have a high index of suspicion. Awareness of the early signs of WE is essential to prevent clinical progression, as patients with the full triad already have a profoundly thiamine-deficient state. This review highlights the neuro-ophthalmic manifestations of WE to guide the clinician in identifying the condition. In addition, we provide an update regarding the clinical characteristics, pathophysiology, neuroimaging and laboratory findings, treatment options, and prognosis of WE.
Introduction
Wernicke encephalopathy (WE) is a life-threatening but reversible neurologic disorder resulting from acute thiamine deficiency. Traditionally, it has been characterized by a clinical triad of altered mental status, ataxia, and ocular dysfunction. This last group of clinical abnormalities are primarily in the purview of the neuro-ophthalmologist. WE was initially described in 1881 by Carl Wernicke, a 33-year-old German physician, who had immersed himself in the developing field of neuroscience. He reported three patients with WE, including two men (aged 33 and 36 years) who consumed large quantities of alcohol and one woman (aged 20 years) who ingested sulfuric acid, leading to pyloric stenosis.Citation1,Citation2 All three patients had ocular motor abnormalities. Wernicke performed an autopsy on each patient, providing clinical-pathological correlation. Six years later, Sergei Korsakoff, a Russian neuropsychiatrist, documented a unique amnestic syndrome, which he called psychosis polyneuritica, but now bears his name, Korsakoff Syndrome (KS).Citation3 In 1897, Murawieff postulated a common etiology for both WE and KS.Citation4 However, it was not until the 1940s that the underlying cause was found to be thiamine deficiency. Campbell and Russell were among the earliest to suggest the association in their case series of 21 patients with WE.Citation5 They noted that only five of their patients abused alcohol which, at that time, was believed to be the likely etiology. Other comorbidities included gastric carcinoma, pyloric stenosis, bowel resection, and severe vomiting. These variable causes led Campbell and Russell to write
… the probability, or at least possibility, of deficient vitamin intake or absorption is obvious from the nature of the primary condition of which encephalopathy is a complication, and the frequency of an accompanying polyneuritis strongly incriminates vitamin B1.Citation5
This review will provide the reader with an overview of WE, highlighting its neuro-ophthalmic involvement. It behooves all clinicians to have a high index of suspicion for WE in the appropriate clinical setting. Prompt diagnosis and treatment can avoid permanent neurologic and ophthalmic sequelae and even death, which can occur if the diagnosis is overlooked and left untreated.
The Clinical Syndrome
While the presentation of WE is highly variable, description of the symptom triad is the traditional starting point (). Yet the complete triad is only seen in 10–33% of patients,Citation6–Citation8 since the complete triad represents an advanced thiamine-deficient state.Citation9 An estimated 75–80% of WE cases are misdiagnosed based on postmortem necropsy studies.Citation6,Citation10 There is no single diagnostic study used to identify the disease. Rather, it is a clinical diagnosis.Citation11
Table 1 Diagnostic Triad of Wernicke Encephalopathy
The estimated prevalence of WE is 0.04–0.13% based on clinical studies but autopsy studies suggest a true prevalence of ~0.4–2.8%.Citation6,Citation7,Citation10 WE is believed to be more common in men than women with a ratio of 1.7 to 1.Citation6 The prevalence of WE in children appears to be similar to adults. There is substantial variability in prevalence of WE in different geographical locations primarily due to the dietary habits of particular populations.
Encephalopathy
Altered mental status is the most common manifestation of the WE triad,Citation12,Citation13 occurring in all but 10% of patients.Citation14 Mental changes can range from apathy to more severe involvement including obtundation and, rarely, coma.Citation8 In approximately 80% of cases, if left untreated, WE transitions into Korsakoff Syndrome (KS), which is characterized by chronic memory impairment, retrograde or anterograde amnesia, confabulation, and mood disturbance such as indifference or mild euphoria.Citation15
Ataxia
Gait impairment in WE ranges from a mild unsteadiness to a complete inability to stand. WE more commonly causes truncal instability and a wide-based gait rather than appendicular ataxia, which is rare.Citation16 Gait ataxia may be due to preferential damage to the superior cerebellar vermis. Concomitant peripheral neuropathy or vestibular dysfunction also frequently contribute to gait impairment in WE.Citation15
Neuro-ophthalmic Findings
Neuro-ophthalmic manifestations of WE involve both the efferent and afferent visual systems. There is a spectrum of ocular motor abnormalities that occurs in patients with WE. Nystagmus is the most common ophthalmic sign and is typically horizontal gaze-evoked nystagmus (GEN).Citation7,Citation8 Of note, GEN is thought to be the earliest indication of thiamine deficiency.Citation9 Horizontal GEN often evolves in stages in patients with WE. Initially, it manifests as brief, nonsustained nystagmus. Next is sustained nystagmus without deficits in gaze-holding. Finally, the nystagmus is accompanied with gaze-holding failure, indicating involvement of the nucleus prepositus hypoglossi (horizontal gaze neural integrator).
Bilateral abducens palsy is the next most common ophthalmic finding, followed by conjugate gaze palsies, more frequently horizontal than vertical.Citation17,Citation18 Unilateral internuclear ophthalmoplegia has been rarely reported,Citation19 but bilateral is especially rare, and only one case could be found.Citation20 One might expect complete ophthalmoplegia to be common in WE since it is often mentioned as part of the classic triad. Yet this is an infrequent occurrence in WE.Citation18,Citation21,Citation22 The evolution of ocular motility disturbances from the various stages of nystagmus to ophthalmoparesis and ultimately, in advanced disease, complete ophthalmoplegia, has been well documented clinically and supported in an animal model.Citation23
Other abnormalities of the efferent visual system have been described in WE. Bilateral and often severe impairment of the vestibulo-ocular reflex (VOR) is a frequent and early manifestation of gaze-holding failure detected with the head impulse test (HIT).Citation24 A study assessing the utility of video HIT in five patients with WE demonstrated involvement of the horizontal VOR with sparing of vertical VOR due to selective impaired function of the horizontal semicircular canals.Citation24 Similar findings were also seen in a report by Choi et al.Citation25 These studies showed that the selective impairment of the horizontal VOR is due to dysfunction of the medial vestibular nuclei (MVN).
Primary position upbeat nystagmus has also been described in WE.Citation26 In some cases, the upbeat nystagmus switched to permanent downbeat nystagmus.Citation27–Citation31 Upbeat nystagmus has been shown to result from lesions to the caudal medulla in the perihypoglossal region (intercalatus and Roller nuclei), whereas downbeat nystagmus results from lesions to the cerebellar flocculus or paramedian tract neurons.Citation31 After transitioning from upbeat nystagmus, downbeat nystagmus is thought to have a poor recovery and reflect a chronic state, designated as a nonprogressive downbeat nystagmus/truncal ataxia syndrome.
Changes in the appearance of the optic disc and retina characterize involvement of the afferent visual system. Optic disc edema and retinal hemorrhages may occur in WE (), and were described in Wernicke’s original report.Citation1 Nevertheless, optic disc edema is an uncommon finding.Citation32–Citation38 It was found in only 2 of 52 (4%) WE patients reported by De Wardener et al.Citation21 Retinal hemorrhages are also unusual and were seen in only 1 of 52 (2%) WE patients described by De Wardener et al and 6 of 245 (2%) WE patients in a case series by Victor et al.Citation39 One could reasonably speculate that the frequency of fundus abnormalities is underestimated since they could be easily missed in patients evaluated by a physician not skilled in ophthalmoscopy.
Figure 1 Fundus changes in Wernicke encephalopathy. At initial presentation (A), there is bilateral optic disc edema with peripapillary and scattered retinal hemorrhages. At one week (B) and two weeks (C) following thiamine supplementation, there is significant interval improvement. At three weeks (D) following treatment, there is complete resolution of the optic disc edema and retinal hemorrhages except one residual hemorrhage along the inferotemporal arcade of the left eye. Both optic discs became pale with final best-corrected visual acuity of 20/600 in each eye. Reprinted from J Formos Med Assoc, 112(3), Yeh WY, Lian LM, Chang A, Cheng CK. Thiamine-deficient optic neuropathy associated with Wernicke’s encephalopathy in patients with chronic diarrhea, 165–170, Copyright (2013), with permission from Elsevier.Citation36
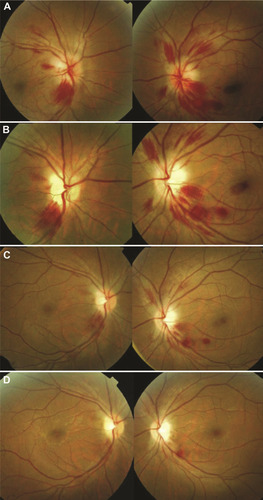
Vision loss is another relatively uncommon finding in WE. A study by Li et al found only 13 published cases with vision loss.Citation35 If it occurs, vision loss is typically severe and bilateral with acuity worse than 20/200 and may lead to loss of light perception.Citation40,Citation41 Vision loss in WE is often due to optic neuropathy with disc edema or associated retinal hemorrhage. However, a few cases of vision loss have been reported even in patients with normal-appearing discs.Citation35
There are reports of other ophthalmic findings in patients with WE. These include altered pupil reactivityCitation15,Citation18,Citation20,Citation42 or size,Citation43–Citation45 light-near dissociation,Citation16,Citation46 impaired convergence,Citation47 spasm of the near reflex,Citation48,Citation49 and ptosis.Citation18,Citation21,Citation39,Citation50 Although ptosis is infrequent in humans, it appears to be common in monkeys with WE, where repeated bouts of thiamine deficiency resulted in increased severity of ptosis.Citation23 Nonetheless, at least two important facts bring into question if the above findings are truly related to WE: (1) physiologic anisocoria is present in approximately 20% of the general population,Citation51 and (2) ptosis may be a manifestation of decreased level of consciousness.
Other Manifestations
Symptoms of irritability, headaches, fatigue, and abdominal discomfort may herald the onset of WE in the setting of subclinical thiamine deficiency.Citation6 Features of hypotension, hypothermia, bradycardia, and respiratory failure can occur from autonomic dysregulation due to lesions in the vagal nuclei or hypothalamus.Citation15,Citation17 Postural hypotension, tachycardia, and electrocardiographic abnormalities may also be seen from cardiovascular dysfunction.Citation14 Mild peripheral neuropathy is common in WE, resulting in paresthesias or burning feet.Citation8 Other uncommon manifestations of thiamine deficiency include: seizures, myoclonus, dysphagia, chorea, dysarthria, orthostatic tremor, and deafness.Citation15 Seizures in WE are thought to result from glutamate toxicity.Citation7
Beriberi
Thiamine deficiency can result in clinical syndromes other than WE. These syndromes fall under the rubric of beriberi. There are two major types of beriberi: wet beriberi which affects the cardiovascular system and dry beriberi which affects the nervous system. Wet beriberi is characterized by high-output, predominantly right-sided heart failure, orthopnea, and pulmonary or peripheral edema.Citation6 Dry beriberi is a painful, distal sensorimotor axonal polyneuropathy that affects the lower limbs more than the upper.Citation47 Patients may experience limb weakness (ie foot drop), decreased proprioception, and reduced or absent peripheral reflexes.Citation50 Gastrointestinal beriberi is a lesser known form which manifests as abdominal pain, nausea, vomiting, and lactic acidosis.Citation7
Infants between two and twelve months of age can develop beriberi if either breastmilk or baby formula is thiamine-deficient.Citation6 Infantile beriberi is characterized by failure to thrive, cardiomyopathy, dyspnea, restlessness, nystagmus, and aphonia. An example of infantile beriberi took place in Israel in 2003. An imported soy-based infant formula was deficient in thiamine, resulting in infantile beriberi in a cluster of newborns.
Korsakoff Syndrome
Early in the disease course, patients with WE may develop memory disturbance, indifference, and disorientation. If left untreated, this impairment can become profound and progress into an amnestic disorder referred to as KS. Rarely, KS occurs in isolation without the preceding acute features of WE.Citation15 Lesions of the dorsomedial thalami have been linked to memory loss.Citation8 An animal study using young chicks found that alcohol neurotoxicity may be a contributing factor to memory loss.Citation52
KS is associated with both anterograde and retrograde amnesia. The former is a defect in acquiring new memories and may be as severe as not recalling events occurring within the past thirty minutes.Citation6 The latter affects the ability to recall events of the recent past, although long-term memory is retained. Working memory rather than reference memory is preferentially impaired. Patients may be rendered capable of only performing the most habitual tasks.Citation14 Memory loss leads to confabulation, which becomes less prominent over time.Citation15 The invented memories fill gaps left by memory loss, and patients believe in the authenticity of the memories. It is thought that these effects on memory are due to irreversible damage to the diencephalic-hippocampal circuits.Citation13
Of note, there are proponents of a unified concept of WE and KS. The neuropathology of the two conditions is very similar. It may be artificial to separate the acute effects of thiamine deficiency as WE with those developing chronically as KS. A more comprehensive designation is Wernicke–Korsakoff syndrome (WKS), comprising a spectrum of clinical findings.
Metabolic Role of Thiamine
Thiamine is a cofactor for several important enzymes in the metabolic pathways supporting cellular function. While thiamine deficiency can affect virtually all organs in the body, its clinical impact is greatest in the brain, heart, and neural tissue.Citation53 Thiamine is a required cofactor for the following enzymes: pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and transketolase.Citation8 The major metabolic pathways involved are shown in . These include the citric acid (Krebs) cycle and pentose phosphate pathway. The citric acid cycle is essential in generating adenosine triphosphate (ATP) and α-ketoglutarate. α-ketoglutarate is then converted to glutamate, an important excitatory neurotransmitter. Acetyl coenzyme A (CoA) is required to initiate the citric acid cycle but also used for the synthesis of acetylcholine and lipids.
Figure 2 The metabolic pathways in which thiamine plays a critical role.
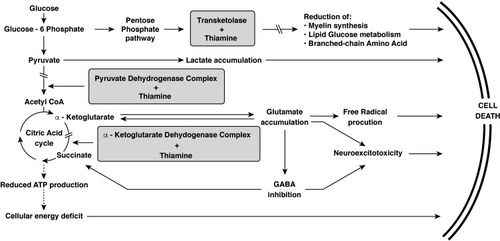
In a thiamine-deficient state, inhibition of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase halts the citric acid cycle, resulting in decreased ATP production.Citation11 This leads to inhibition of the cellular sodium-potassium pumps, resulting in intracellular sodium accumulation and cytotoxic edema, first in astrocytes and then in neural cells with subsequent cell death.Citation30 Another consequence of pyruvate dehydrogenase inhibition is increased intracellular lactate. This low pH state contributes to neuronal necrosis.Citation6 With inhibition of α-ketoglutarate dehydrogenase, there is an increase in intracellular glutamate, which is discharged out of the cell and binds to N-methyl-D-aspartate (NMDA) receptors. This triggers increased intracellular calcium concentrations which, in turn, leads to necrosis and apoptosis.Citation7 Increased intracellular calcium can also induce calcium-dependent nitric acid synthase in the vascular endothelium, disrupt the blood–brain barrier, and result in vasogenic edema.Citation54 Ethanol blocks NMDA receptors at the glutamate site, resulting in receptor upregulation in patients with chronic overuse.Citation55 These patients have increased sensitivity to glutamate. Thus, alcoholism and thiamine-deficiency act synergistically to result in neural excitotoxicity.
The pentose phosphate pathway generates nicotinamide adenine dinucleotide phosphate (NADPH) which is required in the synthesis of nucleic acid and formation of glutathione, a free radical scavenger.Citation7 Inhibition of transketolase leads to accumulation of reactive oxygen species and reduced nucleotide and myelin sheath synthesis.Citation56
Thiamine is absorbed in the small intestine, via an active, carrier-mediated, and rate-limited mechanism, with highest absorption capacity in the duodenum.Citation30,Citation57 In healthy subjects, the maximal amount of thiamine absorption from a single oral administration is approximately 4.5 mg.Citation56 After uptake by the brush border, thiamine is exported out of the enterocyte via the basolateral membrane.Citation55 Most thiamine is protein-bound within the plasma for transport to the tissues.
Humans are incapable of de novo thiamine synthesis and, therefore, require dietary supplementation. The human body can store 30–50 mg of thiamine,Citation7 mainly in the skeletal and cardiac muscle, liver, kidneys, and brain.Citation40 The dietary thiamine requirement is 0.5 mg for every 1000 kcal consumed or about 1.4 mg per day in a person with a typical diet.Citation6 Thiamine deficiency leads to brain lesions within two to three weeks, as body stores of thiamine are only sufficient for 18 days.
Thiamine crosses the blood–brain barrier via both active and passive mechanisms.Citation6 At normal concentrations of thiamine, transport is primarily mediated by an active mechanism. The rate is 0.3 µg/h/g of tissue, which is approximately the normal rate of thiamine turnover in the brain.Citation52 In contrast, high plasma thiamine levels permit rapid passive diffusion across the blood–brain barrier, leading to therapeutic levels in the brain.Citation6,Citation56
Magnesium as a cofactor plays a crucial role in the catalytic activity of many enzymes, including transketolase and thiamine pyrophosphatase.Citation6 The latter is needed for the conversion of thiamine to its active form, thiamine pyrophosphate. This conversion occurs within neural and glial cells at the blood–brain barrier.Citation30 Thiamine pyrophosphate is then used for intracranial metabolism of carbohydrates, lipids, amino acids, and neurotransmitters ().
Genetics
There are a limited number of studies aimed at identifying genetic associations with WE. Evidence that genetic factors play a role is supported by the fact that the Asian population is predisposed to wet beriberi while those of European descent are predisposed to WE or dry beriberi.Citation6 Defects in four genes predispose patients to WE: SLC19A2 or SLC19A3 (encoding proteins for intestinal thiamine transport), SLC25A19 (encoding for thiamine diphosphate transport into the mitochondria), TPK1 (encoding thiamine pyrophosphokinase 1), and TKTL1 (encoding transketolase-like 1).Citation58 Patients harboring these genetic defects are particularly susceptible to WE and require aggressive replenishment of thiamine.
Risk Factors
There is a myriad of risk factors which, by various mechanisms, predispose individuals to WE. These include poor nutrition, diminished gastrointestinal absorption, gastrointestinal loss, increased metabolic demand, and impaired liver metabolism. Alcoholism is the most common risk factor for WE in the industrial world, accounting for approximately 95% of cases.Citation59 Alcohol functions by many of the above mechanisms. Ethanol is toxic to the intestinal mucosa, decreasing thiamine absorption by up to 90%.Citation55 Ethanol causes increased metabolic demand because it is converted to acetyl-CoA, a substrate of the citric acid cycle. Hepatic dysfunction commonly occurs from alcoholism, thereby reducing hepatic thiamine storage. Ethanol also decreases phosphorylation of thiamine to its active form.Citation15
Protracted vomiting of any cause increases the risk of WE. Pyloric stenosis, gastritis, Crohn’s disease, bowel obstruction, pancreatitis, migraine attacks with vomiting, hyperemesis gravidarum, and gastrointestinal or orofacial cancers comprise many of the abdominal conditions that predispose to WE.Citation12 Gastrointestinal surgery is another major risk factor. WE usually develops two to eight months postoperatively in patients with >7 kg of weight loss per month.Citation6 Bariatric surgery has become increasingly popular in recent years with the rising prevalence of obesity.Citation60 Accordingly, the number of published WE cases in the setting of bariatric surgery has increased in recent years.Citation6
Thiamine is mainly derived from foods such as brown rice, cereals, and pork.Citation47 Rice is a predominant part of the diet in about two-thirds of the world’s population. A prime example of unbalanced nutrition is a diet consisting primarily of polished white rice. Because of removal of the husk through milling, polished white rice lacks thiamine.Citation13 The incidence of beriberi in Asian countries has decreased since the initiation of thiamine fortification in their food.Citation6
Other reported causes of thiamine deficiency include hunger strike, religious fasting, anorexia nervosa, total parental nutrition, hypomagnesemia, prolonged diuretic use, hemodialysis, and AIDS.Citation61 Certain foods such as raw fish, shellfish, betel nuts, tea, and coffee contain anti-thiamine agents which may contribute to WE.Citation62 Some diet pills contain herbal properties that inhibit intestinal thiamine absorption. Excessive antacid intake can also impair its absorption.Citation15 Additionally, factors that increase metabolic demand are triggers of WE including: carbohydrate loading, pregnancy, breastfeeding, fever, perioperative period, and Graves’ disease.Citation6,Citation63
Neuropathology
Many patients with WE are not diagnosed until an autopsy is performed because of the variable and, at times, ambiguous clinical presentation. WE has a propensity to affect certain brain areas that have high rates of oxidative metabolism and thiamine turnover.Citation64 In early thiamine-deficient states, vulnerable locations include the nucleus prepositus hypoglossi, MVN, abducens nuclei, and paramedian tract neurons of the pons.Citation9 In later stages of thiamine deficiency, there is involvement of the dorsomedial thalami, mammillary bodies, and cerebral cortex. The periaqueductal gray and superior vermis of the cerebellum appear to be highly thiamine-dependent and are commonly lesioned in WE.Citation64 Involvement can also be seen in the midbrain reticular formation and superior and inferior colliculi.Citation6 The MVN is preferentially sensitive to thiamine deficiency based on an autopsy study that showed pathologic findings in the MVN in 27 of 38 WE cases,Citation65 explaining the high frequency of horizontal VOR impairment in WE. Campbell and Russell observed focal histopathological changes in the optic nerves in two of three patients with WE and vision loss.Citation5
On gross pathology of the brain, about 50% of cases will have bilateral symmetric lesions of grayish discoloration, congestion, and pinpoint hemorrhages. Histological brain examination in the early stage of WE typically demonstrates multiple, symmetric microhemorrhages and spongiosis. Chronic lesions are associated with astrocyte swelling, activated microglia, proliferation of small vessels, and loss of neural or myelinated fibers.Citation30
Ocular motor pathways within the brainstem are especially susceptible to thiamine deficiency. Lesions to cranial nerve nuclei are responsible for the ocular motor palsies.Citation50 However, prior pathologic studies in patients with WE and ophthalmoparesis have shown only infrequent destructive lesions of the ocular motor nuclei.Citation66 This may explain why nystagmus and ocular motor abnormalities are usually reversible with thiamine repletion.
Optic disc edema in WE has been postulated to result from thiamine-induced mitochondrial dysfunction.Citation67 The edema is due to obstruction of axoplasmic flow with subsequent disc hyperemia and retinal hemorrhages.Citation68 With timely treatment, the disc edema resolves and visual function is often preserved.Citation16,Citation38,Citation40,Citation69 However, if there is necrosis of ganglion cells or myelinated nerve fibers,Citation69 there will be permanent vision impairment. The retinal hemorrhages have a predilection for the peripapillary area, and have similar pathologic characteristics to the hemorrhages found in the mammillary bodies in patients with WE.Citation67 Vomiting may also contribute to the development of retinal hemorrhages, although they typically appear in the fovea or parafovea rather than the peripapillary region.Citation16
Diagnostic Evaluation
The initial step in the diagnostic work-up is to suspect WE in patients with any element of the clinical triad, especially in the presence of risk factors such as alcohol abuse. Caine et al devised an operational criteria to assist clinicians in obtaining a diagnosis of WE in alcoholics.Citation70 The criteria requires two of four of the following findings to be present: eye signs, cerebellar dysfunction, altered mental status or mild memory impairment, and dietary deficiencies. This criteria was found to have a 94% sensitivity and 99% specificity for diagnosis of WE. Rapid clinical response to intravenous thiamine repletion can also assist in establishing the correct diagnosis.
Hematologic studies are available to detect thiamine deficiency. Thiamine levels should be measured in the whole blood rather than the plasma since most circulating thiamine is albumin-bound. Free plasma and urinary thiamine concentrations do not accurately reflect tissue concentrations,Citation15 and are consequently tests that lack sensitivity and specificity.Citation8 High-performance liquid chromatography is used for these studies, typically requiring about two to six days for a result. Red blood cell transketolase activity is an alternative test that was previously thought to be the preferred laboratory study for WE. However, Talwar et al have shown that while both whole blood thiamine level and red blood cell transketolase activity are efficacious tests, whole blood thiamine level has practical advantages, such as easier standardization of results and ability to store samples.Citation71 Although laboratory studies have utility, WE cannot be diagnosed solely based on a thiamine level, as there is no critical level below which all patients develop the disorder.
Magnetic resonance imaging (MRI) of the brain is the most valuable test to support a diagnosis of WE because of its high specificity.Citation72 MRI reveals WE-related lesions in one-half to two-thirds of WE patients.Citation8,Citation62 Consequently, a negative brain MRI cannot exclude the diagnosis of WE. Typical MRI findings include bilateral, symmetric T2-weighted, fluid-attenuated inversion recovery (FLAIR), diffusion weighted imaging (DWI), or T1 post-contrast hyperintensities in the medial thalami, mammillary bodies, periaqueductal area, or tectal plate ().Citation7 There are also several regions that atypically demonstrate the above MRI abnormalities including the cerebellum, dorsal medulla, red nuclei or substantia nigra of the midbrain, cranial nerve nuclei, corpus callosum, fornices, head of the caudate nuclei, and cerebral cortex. Cortical MRI lesions suggest permanent damage and are a poor prognostic sign.
Figure 3 Brain MRI findings in Wernicke encephalopathy. Axial views. (A) Precontrast T1 scan is unremarkable. (B) Postcontrast T1 scan demonstrates enhancement of the mammillary bodies. (C) T2 FLAIR image reveals hyperintensity of the periaqueductal gray. (D) Diffusion-weighted imaging shows hyperintense signal in the medial thalami. (E) ADC map shows mild diffusion restriction, consistent with cytotoxic edema.
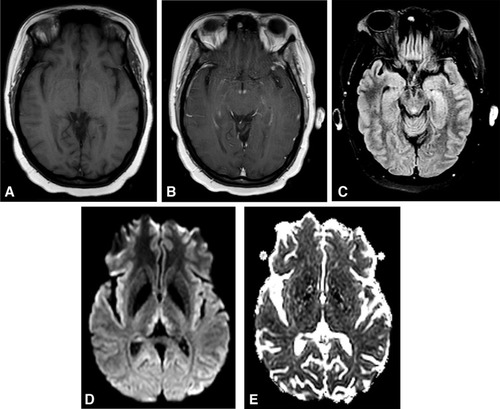
DWI may be consistent with cytotoxic edema (increased DWI and decreased ADC values, ie restricted diffusion) or vasogenic edema (increased DWI and increased or normal ADC values).Citation7 Cytotoxic edema results from dysfunction of cellular osmotic gradients due to thiamine deficiency. Lesions of restricted diffusion indicate tissue at risk, much like ischemic penumbra in stroke. These lesions often regress with administration of thiamine but, in some cases, there is advancement to gliosis, and neurologic deficits may persist.Citation30 The DWI sequence is especially useful for diagnosis in early stages of WE,Citation15 and is more sensitive than T2-weighted imaging at detecting abnormalities.Citation11
Contrast-enhanced T1-weighted images can be particularly valuable in the diagnosis of WE. T1 post-contrast enhancement is identified in about half of WE patients.Citation7 There are reports of mammillary body enhancement as the only imaging abnormality.Citation73,Citation74 T1 post-contrast is also especially useful in the early stages of WE, as it indicates breakdown of the blood–brain barrier.
Computed tomography (CT) of the brain has limited value in acute WE since it is normal in most cases. However, it can demonstrate lesions of decreased signal intensity in the periaqueductal gray or medial thalami.Citation7 Magnetic resonance spectroscopy (MRS) may assist in diagnosis of WE, but its utility is currently undetermined given an insufficient number of published studies. Acute WE findings are associated with increased lactate in the cerebellum and/or decreased N-acetylaspartate (NAA)/creatinine ratio in the thalami and cerebellum.Citation75–Citation77
Differential Diagnosis
Several conditions should be considered in the differential diagnosis of WE. Miller Fisher syndrome has findings that mimic WE including ataxia, areflexia, and ophthalmoplegia.Citation63 However, descending paralysis and a preceding upper respiratory tract infection are distinguishing features. Anti-GQ1b antibodies are present in 90% of cases of Miller Fisher syndrome. Commonly occurring in alcoholics, severe hypophosphatemia has similar clinical characteristics to WE such as altered mental status, peripheral neuropathy, and weakness (including the extraocular muscles).Citation63,Citation78 Central pontine myelinolysis (CPM), which is associated with rapid correction of hyponatremia, is another condition resembling WE that is predominantly observed in alcoholics.Citation63 CPM manifests with altered mental status, diplopia, limb weakness, dysphagia, and dysarthria. Top-of-the-basilar syndrome (ie paramedian thalamic infarction) is another mimic, and patients may present with impaired consciousness and ocular motor abnormalities.
Marchiafava–Bignami is a rare disease associated with alcoholism and vitamin B complex deficiencies that mimics WE.Citation8 MRI abnormalities of the corpus callosum are characteristic of Marchiafava–Bignami and used to differentiate this diagnosis. The differential diagnosis for WE also includes primary CNS lymphoma, Leigh syndrome, variant Creutzfeldt–Jakob disease, paraneoplastic encephalitis, Behçet disease, Whipple disease, multiple sclerosis, alcoholic pellagra encephalopathy, and anoxic encephalopathy.Citation15 It is important to consider alternate causes of encephalopathy such as drug/toxin overdose, sepsis, and delirium tremens. Specifically, acute intoxication from methyl bromide and chronic intoxication from bromvalerylurea can resemble WE.Citation6
While abnormalities on brain MRI can support a diagnosis of WE, they are not pathognomonic. Several other entities may demonstrate symmetric signal alterations of the medial thalami including top-of-the-basilar syndrome, cerebral venous sinus thrombosis, primary CNS lymphoma, acute disseminated encephalomyelitis (ADEM), and viral encephalitis.Citation7
Treatment
WE is a medical emergency and delay in treatment can lead to permanent neurologic sequelae or even death. While there is no doubt that administration of thiamine is critical, the precise dose and route of administration remains controversial. The half-life of thiamine is only 96 minutes, so dosing twice to three times daily is often recommended over administration of a single dose.Citation56,Citation79 There is literature supporting intravenous delivery of thiamine over an intramuscular route.Citation8 Additionally, oral thiamine has been found to be ineffective for both treatmentCitation21 and prophylaxis of WE due to poor gastrointestinal absorption.Citation56
Nevertheless, the guidelines for treatment of WE are unsettled. For example, there are two different clinical protocols in Europe: Royal College of Physicians (RCP)Citation56 and European Federation of Neurological Societies (EFNS).Citation79 However, there has only been one relevant randomized double-blinded clinical trial.Citation79 This clinical trial specifically evaluated the efficacy of 5 to 200 mg once daily doses of intramuscular thiamine in alcoholics and found 200 mg to be optimal.Citation80 Based on Cochrane reviews published in 2004 and 2013 evaluating treatment of alcohol-related WE, there is insufficient evidence from randomized controlled trials to indicate optimal dose, route, frequency, or duration of thiamine treatment.Citation81,Citation82
Thiamine is of limited toxicity with only rare adverse effects. In a study involving 989 patients receiving a total of 1070 doses of 100 mg of intravenous thiamine, 11 developed local irritation and one had generalized pruritus.Citation6 As of 2002, only seven cases of thiamine-related anaphylaxis had previously been reported.Citation56 This lack of thiamine toxicity has enabled high-dose regimens.
Importantly, thiamine should always be given prior to glucose. Glucose supplementation will increase demand on the metabolic pathways that are already deficient in thiamine, precipitating WE in subclinical cases or worsening existing WE.Citation6,Citation8 Similarly, when managing cases of suspected WE, the clinician should be aware of refeeding syndrome, which can occur when replenishing nutrition in the chronically malnourished.Citation41 This typically occurs within four to seven days of refeeding and results in increased glycogen, fat, and protein synthesis.Citation83 With increased metabolic demand, thiamine and magnesium levels may fall, leading to WE. Moreover, because magnesium is a cofactor for thiamine-dependent enzymes, its level must be monitored and supplemented when indicated in all cases of WE.Citation8 Lastly, there are other less established treatments of WE that have shown potential efficacy including donepezil and memantine.Citation55
Prognosis
Delay in diagnosis or inadequate thiamine replenishment greatly affects the clinical outcome in WE. Death has been reported in up to 20% of cases, most often due to pulmonary infection, decompensated liver failure, sepsis, and an irreversible thiamine-deficient state.Citation14
Most neuro-ophthalmic findings of WE are reversible and improve dramatically within the first few weeks of thiamine treatment, since they are not dependent on irreversible pathologic changes.Citation66 Nystagmus improves rapidly within hours of thiamine supplementation in two-thirds of cases.Citation30 Late improvement is seen in most other cases, but there are a few instances of patients with no improvement long-term. Abducens paresis usually completely resolves within days.Citation20 The prognosis for visual recovery in WE is also favorable, but there are reports of permanent visual impairment.Citation35,Citation36,Citation41
Improvement in gait ataxia is common, but in contrast to ophthalmic abnormalities, the recovery is delayed and typically incomplete.Citation8 Most patients are left with residual gait impairment. Mental status changes including drowsiness, disorientation, and apathy gradually improve following thiamine supplementation over the course of many months. However, residual memory and learning deficits frequently persist. The prognosis for recovery is worse in alcoholics since they more likely to ultimately develop KS.Citation7 This is thought to be due to cumulative damage from repeated thiamine-deficient events.
Conclusion
WE is a diagnosis that clinicians cannot afford to overlook. It may manifest as a variable combination of symptoms and signs, often leading to delay in diagnosis and treatment. Even with prompt recognition, the prognosis remains guarded.
A number of challenges remain in expanding our knowledge of WE. Further studies are needed to better understand and effectively reverse neuronal cell death. Randomized controlled trials are needed to provide better guidelines for optimal thiamine treatment. Identification of individuals genetically predisposed to WE could establish the role for prophylaxis of this disorder and reduce both its morbidity and mortality.
Disclosure
The authors report no conflicts of interest in this work.
References
- Wernicke C. Die akute haemorrhagische polioencephalitis superior. Lehrbuch der Gehirnkrankheiten fur Aertze und Studirende. 1881;2:229–242.
- Thomson AD, Cook CC, Guerrini I, Sheedy D, Harper C, Marchall EJ. Wernicke’s encephalopathy revisited. Alcohol Alcohol. 2008;43(2):174–179. doi:10.1093/alcalc/agm14418056751
- Korsakoff SS. Disturbance of psychic function in alcoholic paralysis and its relationship to disturbance in the psychic sphere in multiple neuritis of non-alcoholic origin. Vestn Klin Psichiat Neurol. 1887;4(2):1–102.
- Murawieff W. Zwei fälle von polioencephalitis acuta haemorrhagica superior (Wernicke). Neurol Zentralbl. 1897;16:56–61.
- Campbell ACP, Russell WR. Wernicke’s encephalopathy: the clinical features and their probable relationship to vitamin B deficiency. Q J Med. 1941;10:41–64. doi:10.1093/oxfordjournals.qjmed.a066371
- Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442–455. doi:10.1016/S1474-4422(07)70104-717434099
- Manzo G, De Gennaro A, Cozzolino A, Serino A, Fenza G, Manto A. MR imaging findings in alcoholic and nonalcoholic acute wernicke’s encephalopathy: a review. BioMed Res Intl. 2014;2014:1–12. doi:10.1155/2014/503596
- Sinha S, Kataria A, Kolla BP, Thusius N, Loukianova L. Wernicke encephalopathy – clinical pearls. Mayo Clin Proc. 2019;94(6):1065–1072. doi:10.1016/j.mayocp.2019.02.01831171116
- Kattah JC. Early signs of thiamine deficiency: a case report. Ann Intern Med. 2020. doi:10.7326/L19-0836
- Ogershok PR, Rahman A, Brick J, Nestor S. Wernicke encephalopathy in nonalcoholic patients. Am J Med Sci. 2002;323(2):107–111. doi:10.1097/00000441-200202000-0001011863078
- Chitra S, Lath KV. Wernicke’s encephalopathy with visual loss in a patient with hyperemesis gravidarum. J Assoc Physicians India. 2012;60:53–56.
- Scalzo SJ, Bowden SC, Ambrose ML, Whelan G, Cook MJ. Wernicke-Korsakoff syndrome not related to alcohol use: a systematic review. J Neurol Neurosurg Psychiatry. 2015;86(12):1362–1368. doi:10.1136/jnnp-2014-30959825589780
- Chandrakumar A, Bhardwaj A, Jong GW. Review of thiamine deficiency disorders: Wernicke encephalopathy and Korsakoff psychosis. J Basic Clin Physiol Pharmacol. 2018;30(2):153–162. doi:10.1515/jbcpp-2018-007530281514
- Zubaran C, Fernandes JG, Rodnight R. Wernicke-Korsakoff syndrome. Postgrad Med J. 1997;73(855):27–31. doi:10.1136/pgmj.73.855.279039406
- Kumar N. Neurologic presentations of nutritional deficiencies. Neurol Clin. 2010;28(1):107–170. doi:10.1016/j.ncl.2009.09.00619932379
- Kulkarni S, Lee AG, Holstein SA, Warner JE. You are what you eat. Surv Ophthalmol. 2005;50(4):389–393. doi:10.1016/j.survophthal.2005.04.00515967192
- Reuler JB, Girard DE, Cooney TG. Current concepts. Wernicke’s encephalopathy. N Engl J Med. 1985;312(16):1035–1039. doi:10.1056/NEJM1985041831216063885034
- Deramo VA, Jayamanne DG, Auerbach DB, Danesh-Meyer H. Acute bilateral ophthalmoplegia in a young woman. Surv Ophthalmol. 2000;44(6):513–517. doi:10.1016/s0039-6257(00)00111-910906382
- Winters J, Niespodzany E, Kini TA, Othman BA, Lee AG. Rapid same-day resolution of internuclear ophthalmoplegia in Wernicke encephalopathy following parental high dose thiamine. Can J Ophthalmol. 2020;55(2):e69–e70. doi:10.1016/j.jcjo.2019.07.00731712022
- De La Paz MA, Chung SM, McCrary JA. Bilateral internuclear ophthalmoplegia in a patient with Wernicke’s encephalopathy. J Clin Neuroophthalmol. 1992;12(2):116–120.1629372
- De Wardener HE, Lennox B. Cerebral beriberi: review of 52 cases in a Singapore prisoner-of-war camp. Lancet. 1947;1:11–17. doi:10.1016/S0140-6736(47)91272-520278697
- Donnino MW, Vega J, Miller J, Walsh M. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Ann Emerg Med. 2007;50(6):715–721. doi:10.1016/j.annemergmed.2007.02.00717681641
- Cogan DG, Witt ED, Goldman-Rakic PS. Ocular signs in thiamine-deficient monkeys and in Wernicke’s disease in humans. Arch Ophthalmol. 1960;103(8):1212–1220. doi:10.1001/archopht.1985.01050080124032
- Lee SH, Kim SH, Kim JM, Tarnutzer AA. Vestibular dysfunction in Wernicke’s encephalopathy: predominant impairment of the horizontal semicircular canals. Front Neurol. 2018;9:141. doi:10.3389/fneur.2018.0014129593640
- Choi KD, Oh SY, Kim HJ, Kim JS. The vestibulo-ocular reflexes during head impulse in Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 2007;78(10):1161–1163. doi:10.1136/jnnp.2007.12106117578853
- Sharma S, Sumich PM, Francis IC, Kiernan MC, Spira PJ. Wernicke’s encephalopathy presenting with upbeat nystagmus. J Clin Neurosci. 2002;9:476–478. doi:10.1054/jocn.2002.112112217687
- Shin BS, Oh SY, Kim JS, Lee H, Kim EJ, Hwang SB. Upbeat nystagmus changes to downbeat nystagmus with upward gaze in a patient with Wernicke’s encephalopathy. J Neurol Sci. 2010;298(1–2):145–147. doi:10.1016/j.jns.2010.08.01220832081
- Sakuma A, Kato I, Ogino S, Okada T, Takeyama I. Primary position upbeat nystagmus with special reference to alteration to downbeat nystagmus. Acta Otolaryngol Suppl. 1996;552:43–46.
- Abouaf L, Vighetto A, Magnin E, Nove-Josserand A, Mouton S, Tilikete C. Primary position upbeat nystagmus in Wernicke’s encephalopathy. Eur Neurol. 2011;65:160–163. doi:10.1159/00032432921372575
- Kattah JC. The spectrum of vestibular and ocular motor abnormalities in thiamine deficiency. Curr Neurol Neurosci Rep. 2017;17(5):40. doi:10.1007/s11910-017-0747-928365885
- Kattah JC, Tehrani AS, Du Lac S, Newman-Toker DE, Zee DS. Conversion of upbeat to downbeat nystagmus in Wernicke encephalopathy. Neurology. 2018;91(17):790–796. doi:10.1212/WNL.000000000000638530348852
- van Noort BA, Bos PJ, Klopping C, Wilmink JM. Optic neuropathy from thiamine deficiency in a patient with ulcerative colitis. Doc Ophthalmol. 1987;67(1–2):45–51. doi:10.1007/bf001426963123189
- Vasconcelos MM, Silva KP, Vidal G, Silva AF, Dominigues RC, Berditchevsky CR. Early diagnosis of pediatric Wernicke’s encephalopathy. Pediatr Neurol. 1999;20(4):289–294. doi:10.1016/s0887-8994(98)00153-210328278
- Sparacia G, Banco A, Lagalla R. Reversible MRI abnormalities in an unusual paediatric presentation of Wernicke’s encephalopathy. Pediatr Radiol. 1999;29(8):581–584. doi:10.1007/s00247005065210415181
- Li JM, Rucker JC. Irreversible optic neuropathy in wernicke encephalopathy and leber hereditary optic neuropathy. J Neuroophthalmol. 2010;30(1):49–53. doi:10.1097/WNO.0b013e3181ce80c620182208
- Yeh WY, Lian LM, Chang A, Cheng CK. Thiamine-deficient optic neuropathy associated with Wernicke’s encephalopathy in patients with chronic diarrhea. J Formos Med Assoc. 2013;112(3):165–170. doi:10.1016/j.jfma.2012.10.01023473530
- Lawton AW, Frisard NE. Visual loss, retinal hemorrhages and optic disc edema resulting from thiamine deficiency following bariatric surgery complicated by prolonged vomiting. Oschner J. 2017;17(1):112–114.
- Palakkuzhiyil N, Rehiman S, Manoj PPB, Hameed S, Uvias NA. Visual loss and optic neuropathy associated with Wernicke’s encephalopathy and hyperemesis gravidarum. J Fam Med Prim Care. 2019;8(3):1243–1245. doi:10.4103/jfmpc.jfmpc_121_19
- Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome: a clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemp Neurol Ser. 1971;7:1–206.5162155
- Gratton SM, Lam BL. Visual loss and optic nerve head swelling in thiamine deficiency without prolonged dietary deficiency. Clin Ophthalmol. 2014;8:1021–1024. doi:10.2147/OPTH.S6422824899800
- Sura AA, Cure JK, Kline LB. Visual loss as a presenting feature of Wernicke encephalopathy. J Neuroophthalmol. 2019;39(3):380–382. doi:10.1097/wno.000000000000077130829946
- Cox TA, Corbett JJ, Thompson HS, Lennarson L. Upbeat nystagmus changing to downbeat nystagmus with convergence. Neurology. 1981;31(7):891–892. doi:10.1212/wnl.31.7.8917195514
- Shorey J, Bhardwaj N, Loscalzo J. Acute Wernicke’s encephalopathy after intravenous infusion of high-dose nitroglycerin. Ann Intern Med. 1984;101(4):500. doi:10.7326/0003-4819-101-4-5006433765
- Halavaara J, Brander A, Lyytinen J, Setälä K, Kallela M. Wernicke’s encephalopathy: is diffusion-weighted MRI useful? Neuroradiology. 2003;45(8):519–523. doi:10.1007/s00234-003-1043-812861431
- Pacei F, Mullin S, Colombo C, Viganò S, Bet L. A case of Wernicke's encephalopathy due to oesophageal achalasia. Neurol Sci. 2013;34(5):799–800. doi:10.1007/s10072-012-1136-4
- Sia PI, Sia DIT, Crompton JL, Casson RJ. Nerve fiber layer infarcts in thiamine deficiency. J Neuroophthalmol. 2015;35(3):274–276. doi:10.1097/WNO.000000000000024325815858
- Tan TXZ, Lim KC, Chung CC, Aung T. Starvation-induced diplopia and weakness: a case of beriberi and Wernicke’s encephalopathy. BMJ Case Rep. 2019;12(1):e227412. doi:10.1136/bcr-2018-227412
- Thompson RA, Lynde RH. Convergence spasm associated with Wernicke’s encephalopathy. Neurology. 1969;19(7):711–712. doi:10.1212/wnl.19.7.7115815135
- Shabbir S, Tong O, Gluck L, Robbins M. Convergence spasm in Wernicke encephalopathy. Neurohospitalist. 2018;8(1):NP1–NP2. doi:10.1177/1941874417690668
- Decker MJ, Issacman DJ. A common cause of altered mental status occurring at an uncommon age. Pediatr Emerg Care. 2000;16(2):94–96. doi:10.1097/00006565-200004000-0000710784210
- Lam BL, Thompson HS, Corbett JJ. The prevalence of simple anisocoria. Am J Ophthalmol. 1987;104(1):69–73. doi:10.1016/0002-9394(87)90296-03605282
- Thomson AD, Guerrini I, Marshall EJ. The evolution and treatment of Korsakoff’s syndrome: out of sight, out of mind? Neuropsychol Rev. 2012;22(2):81–92. doi:10.1007/s11065-012-9196-z22569770
- Klooster A, Larkin JR, Wiersema-Buist J, et al. Are brain and heart tissue prone to the development of thiamine deficiency? Alcohol. 2013;47(3):215–221. doi:10.1016/j.alcohol.2012.12.01423357554
- Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32(2):200–219. doi:10.1016/j.nbd.2008.08.00518790057
- Thomson AD, Marshall EJ. The natural history and pathophysiology of Wernicke’s encephalopathy and Kosakoff’s psychosis. Alcohol Alcohol. 2006;41(2):151–158. doi:10.1093/alcalc/agh24916384871
- Thomson AD, Cook CC, Touquet R, Henry JA. The royal college of physician’s report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and emergency department. Alcohol Alcohol. 2002;37(6):513–521. doi:10.1093/alcalc/37.6.51312414541
- Spinazzi M, Angelini C, Patrini C. Subacute sensory ataxia and optic neuropathy with thiamine deficiency. Nat Rev Neurol. 2010;6(5):288–293. doi:10.1038/nrneurol.2010.1620308997
- Ortigoza-Escobar JD, Melero-Luis M, Arias A, et al. Treatment of genetic defects of thiamine transport and metabolism. Expert Rev Neurother. 2016;16(7):755–763. doi:10.1080/14737175.2016.118756227191787
- Thomson AD. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff syndrome. Alcohol Alcohol Suppl. 2000;35(1):2–7. doi:10.1093/alcalc/35.supplement_1.211304071
- Moss HE. Bariatric surgery and the neuro-ophthalmologist. J Neuroophthalmol. 2016;36(1):78–84. doi:10.1097/WNO.000000000000033226764529
- Kasmaei HD, Baratloo A, Soleymani M, Nasiri Z. Imaging-based diagnosis of wernicke encephalopathy: a case report. Trauma Mon. 2014;19(4):e17403.25717447
- Jenkins PF. Wernicke encephalopathy. Am Orthop J. 2015;65:104–108. doi:10.3368/aoj.65.1.104
- Porfido D, Guerriero S, Giancipoli G, Vertrugno M, Lefons V, Dicuonzo F. Bilateral sixth nerve palsies as manifestation of Wernicke’s encephalopathy in a patient with refractory vomiting. Eye Brain. 2010;2:95–98. doi:10.2147/EB.S1089928539769
- Kim K, Shin DH, Lee YB, et al. Evolution of abnormal eye movements in Wernicke’s encephalopathy: correlation with serial MRI findings. J Neurol Sci. 2012;323(1–2):77–79. doi:10.1016/j.jns.2012.08.01422940074
- Kattah JC, Guede C, Hassanzadeh B. The medial vestibular nuclei, a vulnerable target in thiamine deficiency. J Neurol. 2018;265(1):213–215. doi:10.1007/s00415-017-8670-129143209
- Cogan DG, Victor M. Ocular signs of Wernicke’s disease. Arch Ophthalmol. 1954;51(2):204–211. doi:10.1001/archopht.1954.00920040206007
- Bohnsack BL, Patel SS. Peripapillary nerve fiber layer thickening, telangiectasia, and retinal hemorrhages in wernicke encephalopathy. J Neuroophthalmol. 2010;30(1):54–58. doi:10.1097/WNO.0b013e3181ceb4d020182209
- Mumford CJ. Papilloedema delaying diagnosis of Wernicke’s encephalopathy in a comatose patient. Postgrad Med J. 1989;65(764):371–373. doi:10.1136/pgmj.65.764.3712608577
- Serlin T, Moisseiev E. Fundus findings in wernicke encephalopathy. Case Rep Ophthalmol. 2017;8(2):406–409. doi:10.1159/00047892428924437
- Caine D, Halliday GM, Krill JJ, Harper CG. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62(1):51–60. doi:10.1136/jnnp.62.1.519010400
- Talwar D, Davidson H, Cooney J, St JO’Reilly D. Vitamin B(1) status assessed by direct measurement of thiamin pyrophosphate in erythrocytes or whole blood by HPLC: comparison with erythrocyte transketolase activity assay. Clin Chem. 2000;46(5):704–710. doi:10.1093/clinchem/46.5.70410794754
- Kim WJ, Kim MM. Wernicke’s encephalopathy presenting with bilateral complete horizontal and downward gaze palsy in a malnourished patient. Korean J Ophthalmol. 2017;31(4):372–374. doi:10.3341/kjo.2017.001428682019
- Shogry ME, Curnes JT. Mamillary body enhancement on MR as the only sign of acute Wernicke encephalopathy. AJNR Am J Neuroradiol. 1994;15(1):172–174.8141051
- Kavuk I, Agelink MW, Gaertner T, et al. Wernicke’s encephalopathy: unusual contrast enhancement revealed by magnetic resonance imaging. Eur J Med Res. 2003;8(11):492–494.14644703
- Murata T, Fujito T, Kimura H, Omori M, Itoh H, Wada Y. Serial MRI and (1)H-MRS of Wernicke’s encephalopathy: report of a case with remarkable cerebellar lesions on MRI. Psychiatry Res. 2001;108(1):49–55. doi:10.1016/s0925-4927(01)00304-311677067
- Maschalchi M, Belli G, Guerrini L, Nistri M, Del Seppia I, Villari N. Proton MR spectroscopy of Wernicke encephalopathy. AJNR Am J Neuroradiol. 2002;23(10):1803–1806.12427642
- Rugilo CA, Uribe Roca MC, Zurru MC, Capizzano AA, Pontello GA, Gatto EM. Proton MR spectroscopy in Wernicke encephalopathy. AJNR Am J Neuroradiol. 2003;24(5):952–955.12748100
- Subramanian R, Khardori R. Severe hypophosphatemia. Pathophysiologic implications, clinical presentations, and treatment. Medicine (Baltimore). 2000;79(1):1–8. doi:10.1097/00005792-200001000-0000110670405
- Galvin R, Bråthen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA; EFNS. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010;17(12):1408–1418. doi:10.1111/j.1468-1331.2010.03153.x20642790
- Ambrose ML, Bowden SC, Whelan G. Thiamin treatment and working memory function of alcohol-dependent people: preliminary findings. Alcohol Clin Exp Res. 2001;25(1):112–116. doi:10.1111/j.1530-0277.2001.tb02134.x11198705
- Day E, Bentham P, Callaghan R, Kuruvilla T, George S. Thiamine for Wernicke-Korsakoff syndrome in people at risk from alcohol abuse. Cochrane Database Syst Rev. 2004;1:CD004033. doi:10.1002/14651858.CD004033.pub2
- Day E, Bentham P, Callaghan R, Kuruvilla T, George S. Thiamine for Wernicke-Korsakoff syndrome in people at risk from alcohol abuse. Cochrane Database Syst Rev. 2013;7:CD004033. doi:10.1002/14651858.CD004033.pub3
- Mehanna HM, Moledina J, Travis J. Refeeding syndrome: what it is, and how to prevent and treat it. BMJ. 2008;336(7659):1495–1498. doi:10.1136/bmj.a30118583681