 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
The aim of this study was to improve the physicochemical properties and bioavailability of poorly water-soluble sirolimus via preparation of a solid dispersion of nanoparticles using a supercritical antisolvent (SAS) process.
Methods
First, excipients for enhancing the stability and solubility of sirolimus were screened. Second, using the SAS process, solid dispersions of sirolimus-polyvinylpyrrolidone (PVP) K30 nanoparticles were prepared with or without surfactants such as sodium lauryl sulfate (SLS), tocopheryl propylene glycol succinate, Sucroester 15, Gelucire 50/13, and Myrj 52. A mean particle size of approximately 250 nm was obtained for PVP K30-sirolimus nanoparticles. Solid state characterization, kinetic solubility, powder dissolution, stability, and pharmacokinetics were analyzed in rats.
Results
X-ray diffraction, differential scanning calorimetry, and high-pressure liquid chromatography indicated that sirolimus existed in an anhydrous amorphous form within a solid dispersion of nanoparticles and that no degradation occurred after SAS processing. The improved supersaturation and dissolution of sirolimus as a solid dispersion of nanoparticles appeared to be well correlated with enhanced bioavailability of oral sirolimus in rats. With oral administration of a solid dispersion of PVP K30-SLS-sirolimus nanoparticles, the peak concentration and AUC0→12h of sirolimus were increased by approximately 18.3-fold and 15.2-fold, respectively.
Conclusion
The results of this study suggest that preparation of PVP K30-sirolimus-surfactant nanoparticles using the SAS process may be a promising approach for improving the bioavailability of sirolimus.
Introduction
Sirolimus, a triene macrolide antibiotic with immunosuppressive activity, was isolated from Streptomyces hygroscopicus in 1978.Citation1,Citation2 The formulation of an oral dosage form of sirolimus is challenging owing to its poor water solubility, which is 2.6 μg/mL at 25°C.Citation3 In addition, sirolimus is very unstable in phosphate-buffered saline and HEPES buffer under all conditions; their degradation effect on the drug is slower at 4°C–8°C and fastest at 37°C, at which temperature almost all of the drug is destroyed within 24 hours.Citation4 To improve the solubility of sirolimus, various oral formulations, such as inclusion complexes,Citation5,Citation6 liposomes,Citation7 nanocrystals,Citation8,Citation9 and solid dispersionsCitation10 with polyethylene glycol (PEG), have been studied.
In the field of pharmaceutical technology, solid dispersions are commonly used for solving problems related to poor water solubility and poor bioavailability of active ingredients, as well as instability and dosing problems.Citation11,Citation12 The selection of a carrier and the method of preparation have important influences on the properties of the resulting solid dispersion. Solid dispersions can be prepared by fusion, solvent, and solvent-fusion methods. The most relevant technologies for the manufacture of solid dispersions are vacuum drying, spray drying, freeze drying, hot spin mixing, roll-mixing, co-milling, and spraying on sugar beads using a fluidized bed coating system for removal of organic solvents.Citation13 Despite the great potential of solid dispersion systems for improving drug dissolution, the methods traditionally used for their preparation have serious limitations, often involving physical instability of the solid dispersions during storage, problems with grinding, or difficult removal of solvents.Citation14–Citation17 To address these issues, technology using supercritical fluids as an alternative solvent-evaporation method has been proposed to prepare solvent-free solid dispersions.Citation18–Citation20 Supercritical fluid technology represents a new and interesting route for particle formation that avoids most of the drawbacks of the transitional methods.Citation20
A supercritical fluid can be defined as a substance existing as a single fluid phase above its critical temperature and pressure. Among the supercritical fluids, CO2 has been chosen based on its nontoxic, nonflammable, and inexpensive nature, its relatively high dissolving power, and its relatively low critical parameters (Tc = 31.1°C, Pc = 73.8 bar).Citation22 These properties make CO2 particularly attractive for processing heat-sensitive pharmaceuticals. The supercritical fluid process offers an alternative single-step method of particle formation capable of producing crystalline powders of microparticles or even nanoparticles with a narrow size distribution containing a minimum level of residual solvent. Depending on the specific process configuration, several additional advantages can exist, including high product purity, controlled crystal polymorphism, possible processing of thermolabile molecules, single-step process, easy downstream processing, and environmentally acceptable technology.Citation23
The aim of this study was to develop solid dispersion nanoparticles for enhancement of the physicochemical properties and bioavailability of poorly water-soluble sirolimus, using the supercritical antisolvent (SAS) process. First, excipients for enhancing the stability and solubility of sirolimus were screened. Second, solid dispersions of sirolimus nanoparticles were prepared by the SAS process. Their physicochemical properties in the solid state were characterized by differential scanning calorimetry, powder X-ray diffraction, and scanning electron microscopy. Kinetic solubility, dissolution, stability, and pharmacokinetics in rats were analyzed.
Materials and methods
Sirolimus was obtained from Beijing Everbright Science and Trading Co (Beijing, China; purity, 99.4%). The CO2 with high purity of 99.99% was supplied by Hanmi Gas Co Ltd (Seoul, Korea). Everolimus (purity, 95.9%) and zinc sulfate were purchased from Sigma-Aldrich (St Louis, MO). All organic solvents were high-pressure liquid chromatography (HPLC) grade. All other chemicals were reagent grade. Excipients used were hydroxypropyl cellulose (NISSO HPC-SSL and NISSO HPC-SL, Nippon Soda Co, Tokyo, Japan), hydroxypropylmethyl cellulose (HPMC 2910, Shin-Etsu Chemical Co Ltd, Tokyo, Japan), hydroxypropyl-β-cyclodextrin (HP-β-CD, Kleptose® HPB, Roquette, Lestrem, France), polyvinylpyrrolidone (PVP K17, PVP K25, PVP K30 and PVP K90, BASF Co Ltd, Ludwigshafen, Germany), polyvinylpyrrolidone vinyl acetate (PVP VA64, BASF Co Ltd), polyvinyl alcohol PEG graft copolymer (Kollicoat IR, BASF Co Ltd), ethylene oxide propylene oxide block copolymer (Poloxamer 188 and Poloxamer 407, BASF Co Ltd), lauryl macrogol 32 glyceride (Gelucire 44/14, Gattefossè, Saint-Priest, France), stearoyl macrogol glyceride (Gelucire 50/13, Gattefossè), sucrose distearate (Sucroester 11, Gattefossè), sucrose monopalmitate (Sucroester 15, Gattefossè), d-α-tocopheryl polyethylene glycol 1000 succinate (vitamin E TPGS, Eastman Co, Kingsport, TN), Eudragit EPO (Röhm GmbH and Co KG, Sontheim, Germany), PEG 4000, PEG 6000, and PEG 8000, Sigma), polyoxyethylene (40) monostearate (Myrj 52, Sigma), polyoxyethylene (50) monostearate (Myrj 53, Sigma), nicotinamide (Sigma), ascorbic acid (Sigma), citric acid (Sigma), meglumine (Sigma), glycine (Sigma), L-arginine (Sigma), sodium alginate (NSPH and NSPLL, Kibum Food Chemifa Co, Tokyo, Japan) and sodium lauryl sulfate (SLS, Ducsan Co Ltd, Seoul, Korea).
Effect of excipients on solubility of sirolimus
Solubility analysis was performed by adding excess sirolimus (5 mg) to a capped glass vial containing 3 mL of water with 1 mg/mL excipient. The samples were sonicated for 30 minutes at 37°C and placed in a shaking water bath (60 rpm) at 37°C for 3 days, which had been previously determined to be an adequate time for equilibration. At the end of this period, almost the entire volume (2 mL) was transferred to a clean tube and centrifuged at 15,000 rpm for 20 minutes at 37°C. The supernatant was filtered through a 0.45 μm glass fiber syringe filter (GMF, Whatman, Clifton, NJ) using a glass syringe. The filtrate was diluted with methanol, and the sirolimus concentration was assayed by HPLC.
Stability of sirolimus in dissolution medium
The chemical stability of sirolimus was studied in several dissolution media commonly used in dissolution testing with a VK 7000 dissolution testing station and VK 750d heater/circulator (Vankel, Cary, NC) at 37°C. Simulated gastric (pH 1.2) and intestinal fluids (pH 6.8) were prepared according to USP XXVIII, and dissolution medium (pH 4.0) consisted of sodium acetate and acetic acid. Stability testing was carried out by adding a stock solution of sirolimus in dimethyl sulfoxide (DMSO) to 900 mL of dissolution medium, which had been equilibrated at the desired temperature for at least 1 hour. The initial concentration of sirolimus was 1 μg/mL, and the DMSO concentration in the final solution was 0.11%. Suitable aliquots were withdrawn at various time intervals, and the drug concentration was assayed by HPLC.
Effect of excipients on stability of sirolimus
To establish which excipients enhanced the stability of sirolimus, stability testing was performed by adding a stock solution of sirolimus in DMSO to a capped glass volumetric flask containing 50 mL of excipient dissolved in pH 1.2 simulated gastric fluid at 1 mg/mL concentration. The initial concentration of sirolimus was 1 μg/mL, and the DMSO concentration in the final solution was 0.11%. The samples were placed in a water bath at 37°C in the dark. Suitable aliquots were withdrawn at various time intervals, and the drug concentration was assayed by HPLC.
HPLC analysis
HPLC analyses of in vitro samples of sirolimus were performed on a Waters™ HPLC system consisting of a pump (Model 600), an autosampler (Model 717 plus), and an ultraviolet detector (Model 486 tunable absorbance detector) using a previously reported method.Citation24,Citation25 A C18 analytic column (Zorbax Eclipse XDB-C18, 5 μm, 4.6 mm × 250 mm, Agilent, Santa Clara, CA) was used at 60°C. The mobile phase was 84% methanol and 16% water. The injection volume was 20 μL or 50 μL, and the eluent flow rate was 1.0 mL/minute. The signal was monitored at 278 nm.
HPLC analyses of degradation products of sirolimus were performed on a Waters™ HPLC system consisting of a pump (Model 600), an auto-sampler (Model 717 plus), and an ultraviolet detector (Model 486), and a Shimadzu HPLC system (Kyoto, Japan) consisting of a LC-10ADvp pump, SIL-10A autosampler, and SPD-10ADvp UV detector using a previously reported method.Citation26 A Supelcosil (Supelco, Bellefonte, PA) LC-18 column (4.6 mm × 250 mm, 5 μm) was used. Separation of sirolimus and degradation products was achieved with a linear gradient from 65% to 86% methanol in 5 mM ammonium acetate over 80 minutes at flow rate of 0.5 mL/minute. The injection volume was 50 μL or 100 μL, and the signal was monitored at 276 nm.
Preparation of sirolimus solid-dispersion nanoparticles
The SAS apparatus comprised the following components: two high pressure pumps; circulator and cooling lines for keeping the CO2 pump head and CO2 which flowed out of storage tank cold (−10°C); a prevessel with an electric heating jacket; a precipitation vessel (1.9 L) with a water jacket and separator vessel (0.6 L); and two manual back pressure regulators (Model 26-1723-24-194; Tescom). Further information on the SAS apparatus has been published elsewhere.Citation27 First, sirolimus and excipients were dissolved in organic solvents. Second, CO2 from a storage tank was delivered to the top of the particle precipitation vessel through the outer capillary of the two-flow spray nozzle at a constant rate, using a homemade plunger pump, until the desired pressure was obtained. After the pressure and temperature had equilibrated, the drug solution was introduced into the particle precipitation vessel by an HPLC liquid pump (Model 307; Gilson Inc, Middleton, WI) with supercritical CO2 through the inner capillary of the two-flow spray nozzle. The residual solvent (supercritical CO2 and organic solvents) was drained out of the particle precipitation vessel by the back pressure regulator. At the washing step, additional supercritical CO2 continued to flow into the precipitation vessel to wash out the residual organic solvents solubilized in the SAS. The precipitation vessel was slowly depressurized to atmospheric pressure, and finally, the particles were collected from the internal basket of the precipitation vessel (retained by a metal frit and paper filter).
In vitro characterization of sirolimus solid dispersion nanoparticles
The drug content in the processed samples was determined by HPLC. A sample of approximately 20 mg was dissolved in 100 mL of methanol or methanol/dichloromethane (1:1). Analysis of the residual solvents was carried out on a Shimadzu 2010 model gas chromatograph equipped with a flame ionization detection system. A DB-5 capillary column (30 m × 0.32 mm ID; film thickness, 0.25 μm) was used. The samples were dissolved in dimethylformamide. Butanol was used as an internal standard. Quantification was performed using a calibration curve.
Particle morphology was observed using a scanning electron microscope (JSM-7000F; Jeol Ltd, Tokyo, Japan). Samples were coated with gold and palladium using a vacuum evaporator and were examined at an accelerating voltage of 5 kV.
The particle size and particle size distribution of samples were determined by dynamic light scattering using an electrophoretic light scattering spectrophotometer (ELS-8000; Otsuka Electronics, Hirakata, Japan). The samples were dispersed in mineral oil (Macrol 52; Exxon Mobil Co, Baltimore, MD) and sonicated for 10 minutes at 120 W (Branson 8210; Branson Ultrasonics Co, Danbury, CT). We obtained information about particle size by analyzing dynamic light scattering data using the cumulant method.
Powder X-ray diffraction patterns were recorded on a Rigaku powder X-ray diffraction system (Model D/MAX-2200; Ultima/PC, Japan) with Ni-filtered Cu-Kα radiation. The samples were analyzed over the most informative range, from 5° to 60° of 2θ. The step scan mode was used with a step size of 0.02° at a rate of 3° per minute.
Differential scanning calorimetry measurements were conducted using a differential scanning calorimetry S-650 (Scinco Co Ltd, Seoul, Korea). Samples of 2–3 mg were accurately weighed and sealed in aluminum. The measurements were performed under nitrogen purge over 20°C–250°C at a heating rate of 5°C/minute. An empty pan was used for reference, and the differential scanning calorimetry baseline, temperature, and enthalpy were calibrated with indium before each experiment, at a heating rate of 5°C/minute. A nitrogen flow rate of 20 mL/minute was used for each differential scanning calorimetry run.
The specific surface area of the samples was determined by nitrogen adsorption using a surface area analyzer (ASAP 2010; Micromeritics Instrument Corporation, Atlanta, GA).
For kinetic solubility studies, excess solid (approximately 20 mg of sirolimus) was placed in 100 mL of degassed water in a glass flask at 37°C ± 0.1°C. The samples were placed in a shaking water bath at 60 rpm and 37°C ± 0.1°C. Suitable aliquots were withdrawn at various time intervals using a glass syringe. Approximately 2 mL aliquots were transferred to clean tubes and centrifuged at 15,000 rpm for 10 minutes at 37°C. The supernatants were filtered through a 0.45 μm glass fiber syringe filter (GMF, Whatman) and diluted with methanol. The concentration of sirolimus was determined by HPLC.
Dissolution studies were performed according to the USP XXVIII paddle method using a VK 7000 dissolution testing station and VK 750d heater/circulator (Vankel). The stirring speed was 50 rpm, and the temperature was maintained at 37°C ± 0.1°C. Each test was carried out in 900 mL of degassed water. Accurately weighed samples containing the equivalent of 2 mg sirolimus were placed in the dissolution medium. Then, 4 mL aliquots were withdrawn at various time intervals and filtered using a 0.45 μm GMF filter. At each sampling time, an equal volume of the test medium was replaced. Filtered samples were appropriately diluted with methanol, and the drug concentration was assayed by HPLC.
Sirolimus powder or solid dispersion nanoparticles were spread uniformly on a glass plate to allow uniform irradiation. The samples were positioned 30 cm below the ultraviolet lamps (UVA 315–400 nm, 89.2 W; UVB 280–315 nm, 6.5 W; Ultra Vitalux® and Eversun®, Osram, Munich, Germany) inside a stability chamber to protect the samples from extraneous light, and exposed to ultraviolet irradiation for 0–48 hours at 25°C ± 2°C and 60% ± 5% relative humidity. Samples were taken at 12, 24, 36, and 48 hours, and the photodegradation of sirolimus in each sample was monitored using HPLC.
To evaluate the stability of the sirolimus solid dispersion nanoparticles, each sample was placed in an airtight brown vial and stored at 25°C ± 2°C and 60% ± 5% relative humidity. The powder dissolution in water and X-ray diffraction patterns of each sample were examined periodically.
In vivo characterization of sirolimus solid dispersion nanoparticles
Male Sprague-Dawley rats weighing 200–220 g were obtained from Samtaco Bio Korea Inc (Osan, Korea). All rats had free access to tap water and a pelleted diet. The rats were housed in cages and maintained on a 12-hour light/dark cycle at room temperature (25°C) and 55% ± 10% relative humidity. General and environmental conditions were strictly monitored. The study protocol was approved by the ethics committee of Chungnam National University. All animal experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals at Chungnam National University.
The rats were deprived of food for 24 hours before the experiment, and food was reoffered at 4 hours post-dosing. After anesthesia with diethylether, the femoral artery was cannulated with a 23 gauge polyethylene cannula. The cannula was flushed with 0.3 mL of heparin (50 IU) saline solution to prevent blood clotting. After the rats had recovered from anesthesia, sirolimus powder and solid dispersion nanoparticles were administered by oral gavage at a sirolimus dose of 10 mg/kg. Each formulation was dispersed in 5 mL/kg (0.2% w/v) aqueous methylcellulose immediately before dosing. Serial blood samples (approximately 500 μL each) were collected from the femoral artery before dosing and at 20 and 40 minutes, and 1, 1.5, 2, 3, 5, 8, and 12 hours post-dosing. Blood samples were transferred to individual Eppendorf tubes containing 20 μL of ethylenediamine tetra-acetic acid (2.5% w/v) and stored at −80°C until analyzed. Liquid chromatography-mass spectrometry (LC-MS) analysis was performed within one week after blood sampling.
The sirolimus concentration in the blood samples was determined by LC-MS as reported previously, with slight modifications.Citation28–Citation30 Whole-blood samples (400 μL) were extracted by vortex mixing for 60 seconds with 400 μL of methanol, 400 μL of 6.25% (w/v) zinc sulfate, and 40 μL of internal standard solution (1 μg/mL everolimus in 50% methanol). Precipitated materials were separated by centrifugation (13,000 rpm for 10 minutes at 1°C), and the supernatants were transferred to clean tubes. NaOH (100 μL, 0.1 M) was added to each tube, followed by vortex mixing. Next, 1-chlorobutane was added to the sample tubes. The phases were separated by centrifugation (13,000 rpm for 10 minutes at 1°C), and the organic phase was removed and evaporated to dryness under nitrogen at 40°C for 10 minutes. The residue was reconstituted in 100 μL of 70% methanol, and 10 μL of each sample were analyzed by LC-MS. The LC-MS system consisted of an LC-10ADvp pump, SIL-10A autoinjector, SPD-10ADvp ultraviolet detector, and LCMS-2010A mass spectrometer. Sirolimus was analyzed on a Supelcosil LC-18 column (150 × 4.6 mm, 3 μm) maintained at 60°C, with Supelguard LC-18 (2 cm cartridge) as a precolumn. The mobile phase was 70% acetonitrile and was pumped at a flow rate of 1.0 mL/minute. The detection wavelength was 276 nm. The mass spectrometer was operated in positive ion mode and was connected to the chromatographic system using an atmospheric pressure ionization electrospray interface. [M-Na+] m/z 936.00 for sirolimus and [M-Na+] m/z 980.00 for everolimus were selected as the detecting ions, respectively. The mass spectrometry operating conditions were optimized as follows: drying gas, 1.5 L/minute; CDL temperature, 250°C; block temperature, 200°C; and probe voltage, +4.5 kV. Data processing was performed using LC-MS Solution software (Shimadzu). Sirolimus blood concentrations were quantified by comparison with standard curves generated from spiked blood samples extracted in the same manner as for the test samples. Standard curve samples (5–500 ng/mL) were prepared fresh on each day of analysis and were linear over the range tested, with R2 > 0.99.
Pharmacokinetic data analysis
The AUC0→12h and mean residence time were calculated using noncompartmental analysis (WinNonlin 2.1; Pharsight Corporation, Mountain View, CA). The peak concentration (Cmax) and time taken to reach peak concentration (Tmax) were obtained directly from blood data. To demonstrate statistically significant differences, one-way analysis of variance followed by least-squares difference test and the Student–Newman–Keul test was performed, using SPSS software (v 12.0; SPSS Inc, Chicago, IL).
Results and discussion
Screening of excipients
The excipients were chosen to provide a diversity of structural and physicochemical properties, and were not selected based on any specific hypotheses regarding their likely interactions with the formulation components. The polymers studied included cellulose derivatives (methyl cellulose, HPC, HPMC), PVP, PEG, alginate, and alginate derivatives. Although all the selected surfactants, except SLS, were nonionic, they still represented a wide diversity of functional hydrophilic and hydrophobic groups, as well as surfactant architectures, molecular weights, and melting points. In addition, a hydrotropic agent (nicotinamide), three organic acid/base candidates (citric acid, ascorbic acid, and meglumine), and two sugars (sucrose and mannitol) were studied.
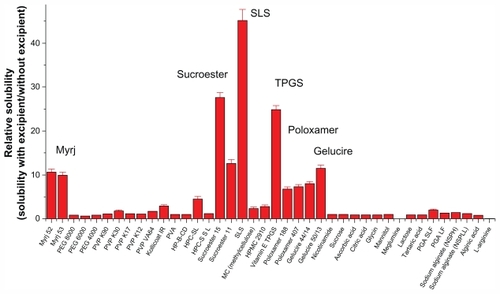
The solubilization effect of the surfactants was greater than that of the polymers, possibly due to the formation of micelles. Each type of surfactant tested had a different solubilization capacity attributable to its particular structure, properties, and interaction with sirolimus. As shown in , the most effective solubilizing excipient was SLS, followed by Sucroester 15, TPGS, Gelucire 50/13, and Myrj 52.
Figure 1 Screening of excipients for improving the solubility of sirolimus.
Note: Data are expressed as the mean ± standard deviation (n = 2–3).
Abbreviations: PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; HPMC, hydroxypropylmethyl cellulose; HPC, hydroxypropyl cellulose; HP-β-CD, hydroxypropyl-β-cyclodextrin; SLS, sodium lauryl sulfate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate; PVA, polyvinyl alcohol; PGA, polyglycolic acid; SL, sirolimus.
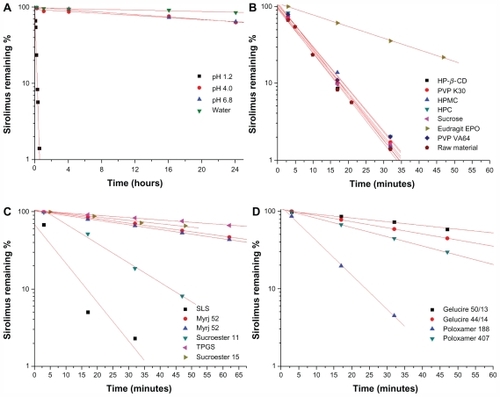
The stability of sirolimus and the effects of excipients on its stability were studied. First, the stability test of sirolimus was performed in different dissolution media using a paddle method as the dissolution apparatus. As shown in , the remaining percentage of sirolimus was decreased in different dissolution media with different degradation kinetics. In particular, rapid degradation of sirolimus was observed in pH 1.2 dissolution medium, and the amount of sirolimus remaining was <10% at 30 minutes. The degradation peaks increased with time. Semilogarithmic plots of the remaining sirolimus versus time in different dissolution media () indicated pseudo first-order degradation behavior:
Figure 2 Stability of sirolimus in dissolution medium (A) and effect of excipients on stability of sirolimus (B, C, and D).
Note: Data are expressed as the mean value (n = 2–3).
Abbreviations: PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; HPMC, hydroxypropylmethyl cellulose; HPC, hydroxypropyl cellulose; HP-β-CD, hydroxypropyl-β-cyclodextrin; SLS, sodium lauryl sulfate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
where [C0] is the initial concentration of sirolimus and [C]t is the percentage remaining at time t, which allowed calculation of the degradation pseudo first-order rate constants (kobs) as the slopes of the lines obtained by linear regression analysis. The half-life (t1/2) was calculated according to Equationequation (2)(2)
The values of kobs and t1/2 are shown in .
Table 1 Stability of sirolimus in dissolution medium
Sirolimus is practically insoluble in water and contains no functional groups that are ionizable in the pH range 1–10.Citation3 Given the rapid degradation of sirolimus in pH 1.2 dissolution medium, the effects of excipients on the stability of sirolimus in pH 1.2 dissolution medium were investigated. The effects of the different excipients on the stability of sirolimus are presented in and . Among the excipients, surfactants (surface-active carriers) enhanced the stabilization of sirolimus to a significantly greater degree than did the hydrophilic polymers. Sucrose and hydrophilic polymers such as PVP, PVP VA64, HPMC, and HPC did not significantly enhance the stability of sirolimus at a concentration of 1 mg/mL. Eudragit EPO, which is soluble at an acid pH, significantly increased the stability of sirolimus, and the pseudo first-order rate constant of sirolimus with Eudragit EPO was decreased approximately four-fold compared with that of sirolimus alone. Previously, Rouf et alCitation5 reported that the stability problems observed for sirolimus in different light, temperature, buffer, and ionic conditions may be avoided by complexation with β-cyclodextrin. However, in the present study, hydroxypropyl β-cyclodextrin did not significantly enhance the stability of sirolimus (1 mg/mL) in pH 1.2 dissolution medium. The stabilization effect of the surfactants was greater than that of the polymers, possibly due to micelle formation, and each surfactant tested had a different solubilization capacity owing to its specific structure, properties, and interaction with sirolimus. The long-chain hydrophobic moiety of surfactants showed a greater stabilization effect than poloxamers and Gelucires. The most effective stabilizer in pH 1.2 dissolution medium was TPGS, followed by Sucroester 15, Gelucire 50/13, and Myrj 52. However, further study of the stabilization mechanism for the excipients is needed.
Table 2 Effect of excipients on stability of sirolimus in a dissolution medium of pH 1.2
Characterization of sirolimus solid dispersion nanoparticles
In preliminary experiments, solid dispersions of sirolimus were prepared using hydrophilic carriers such as HPMC 2910, PVP K30, PVP VA64, HPC-SSL, HPC-SL, PEG 6000, and PEG 8000 at 20% drug:80% polymer (w/w) with ethanol or an ethanol-dichloromethane mixture as a solvent. The operating conditions used in our experiments were selected on the basis of our previous experience with the SAS process. For the particle precipitation vessel, the conditions were 40°C and 12 MPa. In PEG-sirolimus formulations with a drug/polymer ratio of 20%:80%, the yields were low (<10%), and few solid dispersion particles could be collected. In addition, PEG-sirolimus formulations gave irregular microparticles in the size range of 1–20 μm. Furthermore, the PVP VA64-sirolimus formulation resulted in formation of no particles during the SAS process. This might have been attributable to the lower glass transition temperature of PEG 6000 and 8000 and PVP VA64 when using CO2 as a plasticizer.Citation31 Among these hydrophilic polymers, PVP K30 was chosen for further study because it formed a homogeneous matrix.
Effect of surfactants
The experimental conditions and results of the SAS process are summarized in . As shown in , PVP K30-sirolimus nanoparticles with a mean particle size of approximately 400 nm were obtained using the SAS process. Formulations of PVP K30-sirolimus with SLS, or Sucroester 15 produced spherical fine particles. However, when surfactants such as TPGS, Gelucire 50/13, or Myrj 52 were added to the PVP K30-sirolimus formulation, the particle size was increased, and particle aggregation was observed in scanning electron microscopy images (), owing to the lower melting temperatures of these surfactants.Citation32
Table 3 Process conditions and results with the supercritical antisolvent process
Figure 3 Scanning electron microscopic images of raw material (A) and sirolimus solid dispersion nanoparticles prepared by the supercritical antisolvent process: SR-PVP K30 (B), SR-PVP K30-TPGS (C), SR-PVP K30-SLS (D), SR-PVP K30-Sucroester 15 (E), SR-PVP K30-Myrj 52 (F), SR-PVP K30-Gelucire 50/13 (G).
Abbreviations: SR, sirolimus; PVP, polyvinylpyrrolidone; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.

Solid state characterization
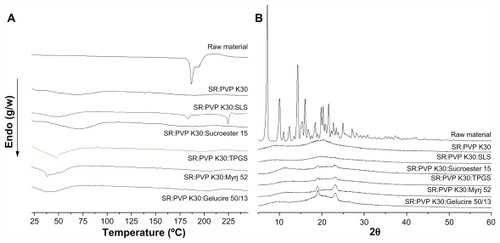
The solid state of sirolimus solid dispersion nanoparticles was characterized by powder X-ray diffraction and differential scanning calorimetry. shows the differential scanning calorimetry thermograms and X-ray diffraction patterns of the raw material and of PVP K30-sirolimus nanoparticles prepared by the SAS process with and without various surfactants. Sirolimus exhibited two endothermic bands around 187°C and 193°C, at a heating rate of 10°C/minute. In contrast, no endothermic peaks corresponding to sirolimus were observed in nanoparticles prepared using the SAS process. The broad endothermic bands observed on the differential scanning calorimetry thermograms of PVP K30 or PVP K30/surfactant were associated with water loss from amorphous PVP K30 and the characteristic melting peaks of surfactants. The diffraction pattern of sirolimus showed characteristic high-intensity peaks at 7.2, 9.9, 10.2, 11.1, 12.5, 14.5, 15.3, 15.5, 16.2, 20.0, 20.4, and 21.8 degrees of 2θ. On the other hand, the powder X-ray diffraction patterns of nanoparticles prepared by the SAS process were completely different from those of the raw material and did not show any characteristic diffraction peaks corresponding to sirolimus. These data confirm that sirolimus was present in an amorphous form within the nanoparticles.
Figure 4 Differential scanning calorimetry thermograms (A) and X-ray diffraction patterns (B) of raw material and sirolimus solid dispersion nanoparticles prepared by supercritical antisolvent process.
Abbreviations: SR, sirolimus; PVP, polyvinylpyrrolidone; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate; SLS, sodium lauryl sulfate.
Kinetic solubility
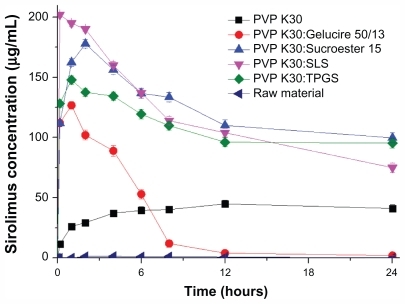
shows the kinetic solubility profiles of the raw material and sirolimus solid dispersion nanoparticles prepared by the SAS process. The maximum solubility of nanoparticles prepared by the SAS process was 40–200 μg/mL, and the solubility at 24 hours was 3–110 μg/mL. In the case of PVP K30-SLS-sirolimus nanoparticles, the maximum solubility of sirolimus was much higher than that of other formulations. However, for PVP K30-Sucroester 15-sirolimus and PVP K30-TPGS-sirolimus nanoparticles, the solubility of sirolimus at 24 hours was much higher than that of other formulations. For supersaturated drugs that precipitate out, the decrease in drug solubility can be prevented by micelle formation with a surfactant. The drug may be molecularly dispersed in the excipient matrix (solid solution), or may occur as fine nanocrystalline or amorphous particles that form during the SAS process. After a solid dispersion formulation (especially solid solution) has dissolved, the drug will be present as a supersaturated solution. Some formulations (polymer and/or surfactant) may serve to inhibit precipitation of the drug from a supersaturated solution, while with other formulations the drug may precipitate out as a metastable polymorph with higher solubility than that of the most stable form.Citation33,Citation34 Therefore, the key factor for an effective solid dispersion formulation is a higher supersaturation concentration for a prolonged time. In the present study, PVP K30-sirolimus nanoparticles containing SLS, TPGS, or Sucroester 15 were effective formulations for a higher supersaturation concentration of sirolimus over a prolonged time.
Figure 5 Kinetic solubility profiles of raw material and sirolimus solid dispersion nanoparticles prepared by the supercritical antisolvent process.
Note: Data are expressed as the mean ± standard deviation (n = 3).
Abbreviations: PVP, polyvinylpyrrolidone; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate; SLS, sodium lauryl sulfate.
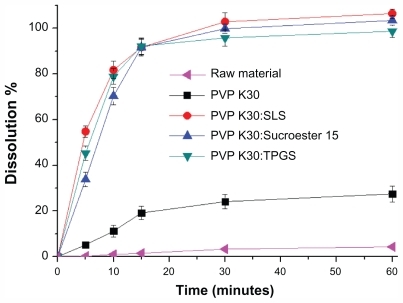
Powder dissolution
The powder dissolution profiles of the raw material and PVP K30-sirolimus with and without surfactants are shown in . The drug was approximately 95% dissolved for PVP K30–sirolimus with surfactants, whereas only approximately 5% of the unprocessed drug was dissolved at 30 minutes. This can be explained by the enhanced supersaturation and an increased specific surface area resulting from reduced particle size of the PVP K30-sirolimus with surfactants in comparison with unprocessed drug.
Figure 6 Powder dissolution profiles of raw material and sirolimus solid dispersion nanoparticles prepared by the supercritical antisolvent process.
Note: Data are expressed as the mean ± standard deviation (n = 3).
Abbreviations: PVP, polyvinylpyrrolidone; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate; SLS, sodium lauryl sulfate.
Pharmacokinetics in rats
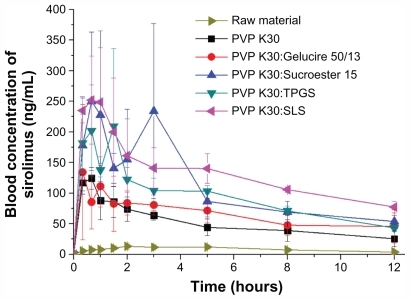
The bioavailability of sirolimus nanoparticle formulations was evaluated in rats. shows the blood concentration- time profile of sirolimus in rats after oral administration of SAS-processed samples at a dose equivalent to 10 mg/kg sirolimus. The pharmacokinetic parameters calculated from the time profiles are presented in . For the raw material (0.2% w/v methyl cellulose as suspension), the AUC0→12h, Cmax, and Tmax were 101.3 ± 14.4 ng · h/mL, 15.9 ± 4.4 ng/mL, and 3.8 ± 1.5 hours, respectively. After the administration of PVP K30-sirolimus nanoparticles, the AUC0→12h, Cmax, and Tmax were 601.7 ± 121.2 ng · h/mL, 129.8 ± 13.9 ng/mL, and 1.0 ± 0.4 hours, respectively. Furthermore, the bioavailability of sirolimus was significantly increased with the addition of surfactants. In particular, PVP K30-SLS-sirolimus nanoparticles showed higher bioavailability than the raw material, with approximately 15.2-fold and 18.3-fold increases in AUC0→12h and Cmax. Based on one-way analysis of variance, there were significant differences among the samples (P < 0.05), which were ranked by the Student–Newman–Keul test in order of increasing AUC0→12h as follows: raw material < PVP K30 and PVP K30-Gelucire 50/13 (P = 0.331) <PVP K30-TPGS, PVP K30-Sucroester 15, and PVP K30-SLS (P = 0.055). In fact, the AUC0→12h and Cmax of sirolimus increased with supersaturation concentration. The bioavailability of PVP K30-sirolimus nanoparticles with surfactants was markedly higher than that of PVP K30-sirolimus nanoparticles without surfactants because of the higher supersaturation concentration over a longer time. The higher supersaturation causes increased flux across biological membranes, saturating P-glycoprotein-mediated efflux and cytochrome P450 3A4 metabolism,Citation35–Citation37 thereby increasing bioavailability of sirolimus. In particular, not only were surfactants such as SLS and Sucroester 15 good excipients for very fine spherical particles in the SAS process, but they also increased the bioavailability of sirolimus due to high supersaturation and rapid dissolution. The results suggest that the preparation of PVP K30-sirolimus-surfactant nanoparticles using the SAS process may be a promising approach for improving the bioavailability of sirolimus.
Table 4 Pharmacokinetic parameters of sirolimus in rats after oral administration of raw material or solid dispersion nanoparticles
Figure 7 Blood concentration-time profile of sirolimus in rats after oral administration of raw material or sirolimus solid dispersion nanoparticles prepared by the supercritical antisolvent process at a dose equivalent to 10 mg of sirolimus/kg of body weight.
Note: Data are expressed as the mean ± standard deviation (n = 4).
Abbreviations: PVP, polyvinylpyrrolidone; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate; SLS, sodium lauryl sulfate.
Photostability test
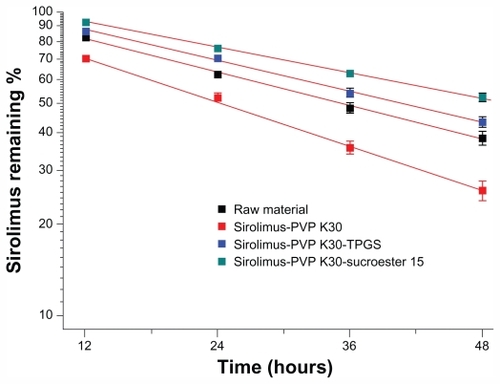
Time-dependent changes in sirolimus content under ultraviolet irradiation were monitored to evaluate whether solid dispersion nanoparticles contribute to protecting sirolimus from photodegradation. Although 38.5% and 25.9% of sirolimus was degraded in raw material and PVP K30-sirolimus nanoparticles, respectively, after 48 hours of ultraviolet irradiation, only 52.3% and 43.5% was degraded in PVP K30-Sucroester 15 and PVP K30-TPGS nanoparticles, respectively (), demonstrating stabilization of sirolimus. The degradation rates of sirolimus were 0.021, 0.028, 0.019, and 0.016 per hour for sirolimus raw powder, PVP K30- sirolimus nanoparticles, PVP K30-Sucroester 15-sirolimus nanoparticles, and PVP K30-TPGS-sirolimus nanoparticles, respectively.
Figure 8 Photodegradation curves of raw material and sirolimus solid dispersion nanoparticles prepared by the supercritical antisolvent process. The samples were exposed to ultraviolet irradiation for 0–48 hours at 25°C ± 2°C and 60% ± 5% relative humidity.
Note: Data are expressed as the mean ± standard deviation (n = 2–3).
Abbreviations: PVP, polyvinylpyrrolidone; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Stability test
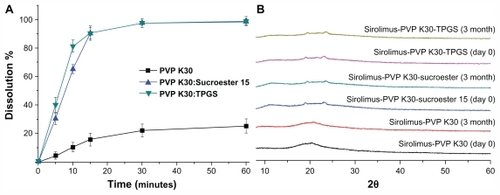
The stability of sirolimus was examined by periodic evaluation of the powder dissolution in water and the X-ray diffraction patterns of PVP K30-sirolimus nanoparticles, PVP K30-Sucroester 15-sirolimus nanoparticles, and PVP K30-TPGS-sirolimus nanoparticles stored at 25°C ± 2°C and 60% ± 5% relative humidity. As shown in , the maximum dissolution percentages of sirolimus from PVP K30-sirolimus nanoparticles, PVP K30-Sucroester 15-sirolimus nanoparticles, and PVP K30-TPGS-sirolimus nanoparticles appeared to be about 25%, 100%, and 100%, and these levels were maintained during 3 months of storage. Furthermore, the X-ray diffraction patterns of solid dispersion nanoparticles were similar on day 0 and after 3 months (). These results indicate that the amorphous state of sirolimus in solid dispersion nanoparticles remained almost unchanged for 3 months. Considering that PVP K30 inhibits recrystallizationCitation38 and Sucroester 15 and TPGS have excellent drug solubilization capacity, the combined use of PVP K30 and Sucroester 15 or TPGS may help to maintain the stability of solid dispersion nanoparticles.
Figure 9 Powder dissolution (A) and X-ray diffraction patterns (B) of sirolimus solid dispersion nanoparticles prepared by the supercritical antisolvent process at day 0 and after 3 months at 25°C ± 2°C and 60% ± 5% relative humidity.
Note: Data are expressed as the mean ± standard deviation (n = 3).
Abbreviations: PVP, polyvinylpyrrolidone; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Conclusion
To improve the oral absorption of sirolimus, a novel sirolimus solid dispersion nanoparticle was designed using the SAS process. PVP K30-TPGS and PVP K30-Sucroester 15 solid dispersion nanoparticles appeared to enhance the supersaturation, dissolution, and stability of sirolimus effectively. Furthermore, pharmacokinetic studies in rats indicated that solid dispersion nanoparticles significantly improved the oral absorption of sirolimus. Overall, the results indicate that PVP K30-TPGS-sirolimus and PVP K30-Sucroester 15-sirolimus solid dispersion nanoparticles demonstrate great potential for clinical application.
Acknowledgments
This work was supported by the Priority Research Centers Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2009-0093815) and, in part, by a grant from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A080470).
Disclosure
The authors report no conflicts of interest in this work.
References
- VézinaCKudelskiASehgalSNRapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principleJ Antibiot (Tokyo)1975287217261102508
- SehgalSNRapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progressionClin Biochem1998313353409721431
- SimamoraPAlvarezJMYalkowskySHSolubilization of rapamycinInt J Pharm2001213252911165091
- RoufMABilensoyEVuralİHincalAADetermination of stability of rapamycin following exposure to different conditionsEuro J Pharm Sci2007321 Suppl 1S46
- RoufMABilensoyEVuralİHincalAAInclusion complexation of rapamycin with beta-cyclodextrin to improve solubility and stability of the drugThe 2nd BBBB Conference on Pharmaceutical SciencesSeptember 13–15, 2007Tallinn-Tartu, Estonia
- BuechGBertelmannEPleyerUSiebenbrodtIBorchertHHFormulation of sirolimus eye drops and corneal permeation studiesJ Ocul Pharmacol Ther20072329230317593014
- AlemdarAYSadleDMcAlisterVCMendezILiposomal formulations of tacrolimus and rapamycin increase graft survival and fiber outgrowth of dopaminergic graftsCell Transplant20041326327115191164
- RosenHAbribatTThe rise and rise of drug deliveryNat Rev Drug Discov2005438138515864267
- JunghannsJUMullerRHNanocrystal technology, drug delivery and clinical applicationsInt J Nanomed20083295310
- PreethamACSatishCSFormulation of a poorly water-soluble drug sirolimus in solid dispersions to improve dissolutionJ Disp Sci Technol201132778783
- VasconcelosTSarmentoBCostaPSolid dispersions as strategy to improve oral bioavailability of poor water soluble drugsDrug Discov Today2007121068107518061887
- LeunerCDressmanJImproving drug solubility for oral delivery using solid dispersionsEuro J Pharm Biopharm2000504760
- SrinarongPde WaardHFrijlinkHWHinrichsWLJImproved dissolution behavior of lipophilic drugs by solid dispersions: the production process as starting point for formulation considerationsExpert Opin Drug Deliv201181121114021722000
- SerajuddinATMSolid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughsJ Pharm Sci1999881058106610514356
- JanssensSVan den MooterGReview: physical chemistry of solid dispersionsJ Pharm Pharmacol2009611571158619958579
- CraigDQThe mechanisms of drug release from solid dispersions in water-soluble polymersInt J Pharm200223113114411755266
- KaravasEGeorgarakisESigalasMPAvgoustakisKBikiarisDInvestigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug-polymer interactionsEuro J Pharm Biopharm200766334347
- WonDHKimMSLeeSParkJSHwangSJImproved physicochemical characteristics of felodipine solid dispersion particles by supercritical anti-solvent precipitation processInt J Pharm200530119920816024189
- JunSWKimMSJoGHCefuroxime axetil solid dispersions prepared using solution enhanced dispersion by supercritical fluidsJ Pharm Pharmacol2005571529153716354397
- JunSWKimMSKimJSPreparation and characterization of simvastatin/hydroxypropyl-beta-cyclodextrin inclusion complex using supercritical antisolvent (SAS) processEur J Pharm Biopharm20076641342117240129
- KalaniMYunusRApplication of supercritical antisolvent method in drug encapsulation: a reviewInt J Nanomed2011614291442
- FagesJLochardHLetourneauJJSauceauMRodierEParticle generation for pharmaceutical applications using supercritical fluid technologyPowder Technol2004141219226
- PasqualiIBettiniRGiordanoFSolid-state chemistry and particle engineering with supercritical fluids in pharmaceuticsEuro J Pharm Sci200627299310
- RicciutelliMDi MartinoPBarboniLMartelliSEvaluation of rapamycin chemical stability in volatile-organic solvents by HPLCJ Pharm Biomed Anal2006411070107416545930
- WangXVenkatramanSSBoeyFYLooJSCTanLPControlled release of sirolimus from a multilayered PLGA stent matrixBiomaterials2006275588559516879865
- PaineMFLeungLYLimHKIdentification of a novel route of extraction of sirolimus in human small intestine: roles of metabolism and secretionJ Pharmacol Exp Ther200230117418611907172
- KimMSJinSJKimJSPreparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) processEur J Pharm Biopharm20086945446518359211
- StreitFChristiansUSchiebelUMMeyerASewingKFStructural identification of three metabolites and a degradation product of the macrolide immunosuppressant sirolimus (rapamycin) by electrospray-MS/MS after incubation with human liver microsomesDrug Metab Dispos199624127212788937863
- MalekiSGravesSBeckerSTherapeutic monitoring of sirolimus in human whole-blood samples by high-performance liquid chromatographyClin Ther200022Suppl BB25B2710823371
- WacherVJSilvermanJAWongSSirolimus oral absorption in rats is increased by ketoconazole but is not affected by D-alpha-tocopheryl poly(ethylene glycol 1000) succinateJ Pharmacol Exp Ther200230330831312235265
- JuppoAMBoissierCKhooCEvaluation of solid dispersion particles prepared with SEDSInt J Pharm200325038540112527165
- KimMSKimJSHwangSJEnhancement of wettability and dissolution properties of cilostazol using the supercritical antisolvent process: effect of various additivesChem Pharm Bull20105823023320118585
- AlonzoDEGaoYZhouDMoHZhangGGZTaylorLSDissolution and precipitation behavior of amorphous solid dispersionsJ Pharm Sci20111003316333121607951
- KonnoHHandaTAlonzoDETaylorLSEffect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipineEur J Pharm Biopharm20087049349918577451
- LampenAZhangYHackbarthIBenetLZSewingKFChristiansUMetabolism and transport of the macrolide immunosuppressant sirolimus in the small intestineJ Pharmacol Exp Ther1998285110411129618413
- PaineMFLeungLYWatkinsPBNew insights into drug absorption: studies with sirolimusTher Drug Monit20042646346715385826
- CumminsCLJacobsenWChristiansUBenetLZCYP3A4-transfected Caco-2 cells as a tool for understanding biochemical absorption barriers: studies with sirolimus and midazolamJ Pharmacol Exp Ther200430814315514569063
- Abu-DiakOAJonesDSAndrewsGPAn investigation into the dissolution properties of celecoxib melt extrudates: understanding the role of polymer type and concentration in stabilizing supersaturated drug concentrationsMol Pharm201181362137121696180