 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Glucagon-like peptide-1 (GLP-1) (7–36) is a peptide incretin hormone released from the endocrine L-cells of the intestinal mucosa with unique antidiabetic potential. Due to low absorption efficiency and instability in the gastrointestinal tract, the introduction of orally active GLP-1 is a large challenge. Here we developed a novel silica-based pH-sensitive nanomatrix of GLP-1 (SPN-GLP-1) in order to provide a strategy for oral peptide delivery.
Methods
SPN-GLP-1 composed of silica nanoparticles and pH-sensitive Eudragit® was prepared and characterized by dynamic light scattering, scanning electron microscope, transmission electron microscope, high-performance liquid chromatography, surface analysis, drug release, and so on. Its permeability across the Caco-2 cell monolayer and intestinal mucosa, proteolytic stability against the intestinal enzymes, pharmacokinetics, hypoglycemic effect in the intraperitoneal glucose tolerance test (IPGTT), and primary toxicity were then evaluated.
Results
It was indicated that the nanomatrix system obtained had a unique nanoscale structure and pH-sensitivity in drug release. It displayed a five-fold intestinal mucosa permeability and significantly higher proteolytic stability compared to native GLP-1 (P < 0.001). A longer half-life was observed after oral administration of SPN-GLP-1, and its relative bioavailability was 35.67% in comparison to intraperitoneal GLP-1. Oral delivery of SPN-GLP-1 significantly reduced the blood glucose level and its hypoglycemic effect over intraperitoneal GLP-1 reached 77%. There was no evident toxicity of SPN-GLP-1 found from both animal status and histochemical analysis of gastrointestinal tissues.
Conclusion
The silica-based pH-sensitive nanomatrix designed and prepared here might be considered as a potential oral delivery system not only for GLP-1, but also for other peptide or macromolecular drugs.
Introduction
Diabetes mellitus is a global disease that increasingly threatens overall human health.Citation1,Citation2 The incretin hormone glucagon-like peptide-1 (GLP-1) is a 3.3 kD MW peptide hormone released from the endocrine L-cells of the intestinal mucosa in response to the ingestion of nutrients. Because of their important roles in glucose metabolism, GLP-1 and its derivatives have been recognized as potent drug candidates for the treatment of type 2 diabetes.Citation3,Citation4 Generally, a long period of drug administration is needed since diabetes is defined as a chronic disease. However, as a macromolecular peptide, GLP-1 is rather poor in permeability across the intestinal epithelium and very vulnerable to enzymatic degradation.Citation5 To overcome those drawbacks, some efforts have been made, such as the development of metabolically stable analogs of GLP-1Citation6,Citation7 and the use of enzyme inhibitors.Citation8 There are limited studies on a drug delivery system for GLP-1, including liposomes and microspheres for prolonged action and enhanced bioavailability.Citation9–Citation11 As a matter of fact, the biotechnology-based macromolecule drugs are in fast development right now, and their oral delivery faces the same challenges as GLP-1.
Nanoscale drug delivery systems have demonstrated outstanding advantages in promoting drug absorption of oral biomacromolecules:Citation12 (1) they provide a huge surface area achieving extremely high dispersion of drug molecules; (2) when the proper polymer is used they offer bioadhesion increasing the interaction between the drug delivery system and mucosa; and (3) they protect biomacromolecules from enzyme degradation. These characteristics are favorable for drug absorption in the gastrointestinal (GI) tract. Actually, great progress has been made, evidenced by the five marketed oral pharmaceutical products and more in clinical trials that use nanotechnology.Citation13 The five oral formulations available in the market, including sirolimus (RAPAMUNE®; Pfizer, Inc, New York, NY), aprepitant (EMEND®; Merck and Co, Inc, Whitehouse Station, NJ), fenofibrate (TriCor®; Abbott Laboratories, North Chicago, IL), megestrol acetate (MEGACE® ES; Par Pharmaceutical Companies, Woodcliff Lake, NJ), and fenofibrate (Triglide™; Sciele Pharma, Atlanta, GA), use nanocrystal technology, which provides drug particles in 100–200 nm.Citation14 This dramatically increases the rate of drug dissolution in vitro, improves oral bioavailability, and reduces variability in absorption and the effect of food. However, challenges remain for nanotechnology-based oral formulation, such as the requirement for sophisticated equipment, high cost, lack of pharmaceutical excipients, the poor long-term stability of the nanoparticles, and the safety of the polymers and surfactants used.Citation15–Citation17 It is worthwhile to mention that most of the successful formulation developments have been related to insoluble drugs but not soluble biomacromolecules.
Currently, strategies to improve the delivery of biomacromolecules including peptides, proteins, and gene drugs mainly focus on their associations with nanocarriers (eg, polymeric nanoparticles).Citation18–Citation20 Insulin nanoparticles for oral delivery were studied most extensively and well viewed.Citation21 However, there is no technology available that can be considered as a breakthrough. Therefore, there is a great need to study more about the nanoscale drug delivery systems for oral delivery of biomacromolecules.
It seems necessary to develop a stable system for biomacromolecules like GLP-1, using only pharmaceutical excipients and a relatively simple preparation process. Keeping these in mind, we designed a nanoscale drug delivery system for the oral delivery of GLP-1 in which the peptide is adsorbed on the surface of a kind of solid nanoparticle and then encapsulated by a pH-sensitive polymer. The solid nanoparticles used here are colloidal silicon dioxide Aerosil® 200 (A200; Degussa, Darmstadt, Germany),Citation22 a pharmaceutical excipient used as a lubricant for many years, and the polymer applied is Eudragit® L100 (Rohm Company, Darmstadt, Germany), which is pH-sensitive so it is possible to protect GLP-1 in acidic conditions like the stomach. And it was shown in our previous study that Eudragit® L100 was able to increase drug adhesion in the GI tract.Citation23 We supposed that such a system can improve GLP-1 absorption in vivo through its effect of high dispersion, protection, and adhesion.
For the proof of concept, a silica-based pH-sensitive nanomatrix (SPN) loaded with GLP-1 (SPN-GLP-1) was prepared and its physicochemical properties were assessed by transmission electron microscopy (TEM), scanning electron microscopy (SEM), dynamic light scattering, high-performance liquid chromatography (HPLC), surface analysis, and an in vitro drug release study. After oral administration, SPN-GLP-1 was evaluated in rats with GLP-1 as the control, in a series of tests including the intraperitoneal glucose tolerance test (IPGTT), pharmacokinetics analysis, and safety study. Moreover, absorption mechanisms such as transcellular and transmucosa transport as well as the proteolytic stability against intestinal enzymes were also investigated.
Materials and methods
Materials
GLP-1 was provided by Novo Nordisk (Copenhagen, Denmark). Eudragit® L100 (Mw 135,000) and SiO2 powder Aerosil® 200 (A200) was kindly provided by Rohm Company and Deggusa Company, respectively. Male Sprague Dawley (SD) rats were purchased from Peking University Experimental Animal Center, Beijing, China and treated at all times according to animal welfare standards similar to Helsinki. A blood glucose reagent kit was obtained from BIOSINO Biotechnology and Science INC (Beijing, China). Unless otherwise specified, all other reagents used were of the highest quality commercially available.
Preparation of SPN-GLP-1
The nanomatrix of GLP-1 with fine silica particles and Eudragit® were prepared using the rotary evaporation method. Briefly, 9 mg A200 silica was suspended in 3 mL distilled water containing 1 mg GLP-1 and dried by vacuum after mixing for 20 minutes under magnetic stirring. Then the solid powder obtained was added into 10 mL ethanol solution containing 10 mg Eudragit® L100 and ultrasonicated for 30 minutes. The ethanol solvent was removed under reduced pressure in a rotary evaporator and finally the solid powder was pulverized and screened.
Characteristics of SPN-GLP-1
Particle size and zeta potential analysis
The particle size and surface charge of SPN-GLP-1 were determined by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at 25°C. The particles were suspended in a large amount of distilled water and analyzed after ultrasonication. Particle size was expressed as volume mean diameter in nanometers.
Morphology and structure
The morphology of SPN-GLP-1 was characterized by a TEM (JEM-1230, JEOL, Tokyo, Japan). For negative staining, one droplet of the sample was diluted 1000-fold with distilled water and placed on the copper grid. Excess fluid was removed using filter paper, and then one drop of 2% uranyl acetate was added 2 minutes before examination with the TEM.
The SPN-GLP-1 was also examined using an SEM (JSM-5600LV; JEOL, Tokyo, Japan). The sample was placed onto an aluminum specimen stub, dried overnight and finally treated by a sputter coating with gold (IB-3 Ion Coater; EIKO, Tokyo, Japan) at 2 mA for 3 minutes prior to imaging.
Surface analysis
The nitrogen adsorption-desorption isotherms were collected at 77 K using an ASAP2010 rapid surface area and pore size analyzer (Micromeritics Instrument Corporation, Norcross, GA) as previously reported.Citation24 Briefly, all samples were degassed at 70°C under a vacuum for 12 hours prior to analysis. The specific surface area and the pore size were determined according to the multiple-point Brunauer–Emmett–Teller method and the Barrett–Joyner–Halenda method using the nitrogen desorption branches of the isotherms. The pore volume was computed based on the Brunauer–Emmett–Teller model.
In vitro release study
HPLC analysis of GLP-1
The high-performance liquid chromatography (HPLC) system used for the analysis of GLP-1 consisting of a SHIMADZU LC-20AT pump, an auto SIL-20 A sampler, a SPD-20A UV detector set at 214 nm, and a 250 × 4.6 mm Ultimate C18 column (Welch Materials Corporation, Ellicott City, MD) at 35°C. The mobile phase consisted of 45% solution A (0.2 M Na2SO4/acetonitrile = 82/18, V/V) and 55% solution B (50% acetonitrile/H2O), and the flow rate was 1.0 mL/minute. The standard curves were linear over the concentration range of 0.1–50 μg/mL.
In vitro release study
An aliquot of SPN-GLP-1 equivalent to 30 mg GLP-1 was suspended in 8 mL pH 1.0 or pH 7.4 phosphate buffered saline (PBS), and shaken in a thermostatic shaker at 37°C for 12 hours at 80 ± 10 rpm. At predetermined time points of 0.5, 1, 2, 3, 5, 7, and 12 hours, a sample of the supernatant was withdrawn and replaced with 0.5 mL of fresh PBS. Then GLP-1 in the supernatant was assayed using the HPLC system mentioned previously.
In vivo evaluation
Animals
Adult male SD rats weighing 180–200 g were purchased from the Laboratory Animal Center of Peking University Health Science Center (Beijing, China). Animals were housed in groups of 3–5 under standard conditions (temperature 22°C ± 2°C, relative humidity 55% ± 5%, 12-hour light/dark cycle) with food and water available ad libitum. All experiments were performed under the guidelines of the Experimental Laboratory Animal Committee of Peking University Health Science Center and were in strict accordance with the principles and guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Hypoglycemic efficacy
The rats were randomly divided into 4 groups (5 rats per group). After fasting for 16–18 hours, the rats were treated with oral SPN-GLP-1, oral GLP-1, intraperitoneal (ip) GLP-1, and oral saline, respectively. SPN-GLP-1 was suspended in water by ultrasonication before test, and the dose of GLP-1 was 1 mg/kg. At the appropriate time after dosing, the animals were given 2 g/kg glucose intraperitoneally. Then, blood samples were drawn from the orbital venous plexus at different intervals and blood glucose levels were measured using a glucose kit (BIOSINO Biotechnology and Science INC, Beijing, China). At the end of the experiments, all rats were sacrificed by anesthetic injection (urethane, 2.5 g/kg). The hypoglycemic degree (Fp% vs ip control group) was calculated as follows:
where AUCGLP-1 is the area under curve of GLP-1 above the 100% horizontal line from 0 minutes to 240 minutes, AUCSPN-GLP-1 is the same value for SPN-GLP-1, Wip means the dose of GLP-1 by intraperitoneal administration, and Wop is the dose of SPN-GLP-1 by oral administration, respectively.
Pharmacokinetics study
Male SD rats in one of 3 groups (5 animals per group) were fasted overnight and received oral SPN-GLP-1, ip GLP-1, or oral saline, respectively. The oral dose and injection dose of GLP-1 was 1 mg/kg and 0.33 mg/kg, respectively. Then, blood samples were taken at 0, 0.25, 0.5, 1, and 2 hours. Serum was obtained and treated as described previously. Samples were analyzed with an ELISA kit (Cusabio Company, Wuhan, China).
Safety evaluation
Male SD rats (200 g) were divided into four groups and treated with different oral formulations: saline, GLP-1 (1 mg/kg), blank SPN, and SPN-GLP-1 (1 mg/kg), respectively. After 24 hours, all rats were sacrificed and different organs and tissues including the GI tract were collected. All of the collected tissue and organs were fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin after a series of dehydration in ethanol and xylene. Specimens were sectioned into 5 mm slices and subsequently stained with hematoxylin and eosin. For each specimen, 10 random slides were obtained and observed. The photos were also taken under the light microscope.
Absorption mechanism study
Proteolytic stabilities
The proteolytic stabilities of SPN-GLP-1 were evaluated using a method described elsewhere,Citation25,Citation26 with slight modifications. Briefly, the intestinal fluid from a 24-hour-prefasted SD rat was obtained by flushing the intestinal part (from the duodenum to end of the jejunum) with 5 mL of 25 mM phosphate buffer (pH 6.4) and centrifuging at 12,000 rpm for 10 minutes. The supernatant was diluted (1:10) with the same buffer. A portion (25 μL) of either a GLP-1 or SPN-GLP-1 solution (200 μg/mL each) was mixed with the same volume of the intestinal fluid (pre-incubated at 37°C for 15 minutes), and incubated at 37°C. After the incubations were stopped by adding 200 μL of 1% trifluoroacetic acid/demineralized water (TFA/DW) in ice-cold methanol, the resulting mixtures were centrifuged at 12,000 rpm for 5 minutes, and the residual GLP-1 in supernatants was analyzed by HPLC.
Transport across Caco-2 cell monolayer
The permeability of SPN-GLP-1 against a biomembrane were examined using a model of a human colon adenocarcinoma (Caco-2) cell line (ATCC, passage number: 30–34). Briefly, the cells were grown until 80% confluence and seeded onto the collagen-coated polycarbonate. Transwell membrane inserts (0.4 μm pore size, 1.13 cm2 area; Corning Costar, Cambridge, MA) at a density of 1.25 · 105 cells/well. The cell monolayer inserts were used when the transepithelial electrical resistance values reached 350–500 Ω · cm2 (after a culture period of 21 days). After washing the cell monolayer with the transport medium (pH 7.4 PBS), 0.5 mL of the transport medium containing the SPN-GLP-1 (5 μM) and 1.5 mL of the drug-free transport medium were added to the apical and basolateral sides, respectively. The drug concentration in the basolateral side was determined using a commercial GLP-1 ELISA kit (Linco Research Inc, St Charles, MO). The transport profiles of GLP-1 from the basolateral to the apical side were examined using the same procedure.
Transport across the mucosa of the intestine
The everted rat gut sac method was used as described previously.Citation27 Briefly, adult male SD rats (200–250 g) were fasted for 24 hours and sacrificed by cervical dislocation. Their small intestines were removed and washed three times with saline at room temperature. Then, the intestine was placed in 37°C and everted. One end was clamped before filling with fresh medium. The intestine was then sealed with a second clamp. Small sacs (2–2.5 cm in length) were then tied using silk braided sutures. The flasks were stoppered with gas-tight silicon bungs. Finally, the resulting gut sacs were incubated at 37°C with SPN-GLP-1 or GLP-1 solution in an oscillating water bath (60 cycles/minute). At the appropriate time points, the sacs were removed, washed four times in saline, and blotted dry. The sacs were cut open and the serosal fluid was collected. Each sac was weighed before and after serosal fluid collection and the area of each sac was measured. The GLP-1 in medium and the serosal fluid was evaluated by HPLC. Then, the absorption amount of GLP-1 in each sac was calculated. Each experiment was carried out using the small intestine from one animal.
Data analysis
Standard noncompartmental pharmacokinetic parameters (± SD) were calculated. Statistical comparisons between groups were made using Microsoft Excel (Microsoft Corporation, Redmond, WA) by the student’s t-test for independent groups assuming equal variances within each group.
Results
Preparation and characterization of SPN-GLP-1
The final SPN-GLP-1 obtained was a kind of solid white powder without any surfactants which often bring up some toxicity, and it is easy to make the solid nanomatrix into a stable solid dosage form like tablets and capsules. The schematic diagram () shows the supposed structure of SPN-GLP-1.
Figure 1 Scheme of the formation of the GLP-1 loaded silica-based pH-sensitive nanomatrix (SPN-GLP-1).
Abbreviations: GLP-1, glucagon-like peptide-1; SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1.
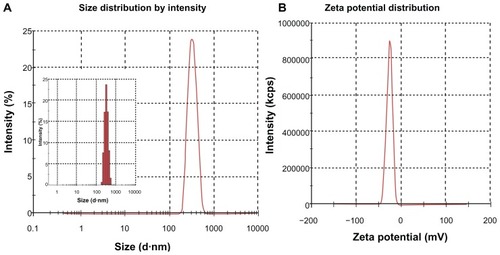
Size and zeta potential
The physiochemical properties of SPN-GLP-1 are listed in . The average particle diameter of SPN-GLP-1 dispersed in water was 307.57 ± 4.45 nm ( and ), and its size distribution was rather narrow as shown by the small PDI value (0.04) and also in . The SPN-GLP-1 had a negative zeta potential of −25.98 ± 1.7 mV ( and ) which is likely related to the structure of Eudragit® L100.
Table 1 Physiochemical properties of silica-based pH-sensitive nanomatrix with GLP-1 (SPN-GLP-1)
Figure 2 (A) Size and (B) zeta potential of SPN-GLP-1 measured by Malvern Zetasizer Nano ZS (Malvern, UK).
Abbreviation: SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1.
Morphology and structure
As seen in , GLP-1 alone had particles with rather large size (in the order of micrometers) while the particles of SPN-GLP-1 were obviously composed of very small particles much less than 50 nm, similar to that of blank SPN without GLP-1. A similar observation of SPN-GLP-1 could be seen in the SEM image (). There were no GLP-1 particles found in the image of SPN-GLP-1 suggesting the highly dispersive state of GLP-1 molecules in the nanomatrix structure. The small nanoparticles deposited together and adhered one by one possibly due to the presence of Eudragit® L100.
Figure 3 (A) Typical scanning electron micrographs of SPN-GLP-1 (scale bar: 1 μm) and (B) transmission electron microscope image of GLP-1, blank nanomatrix, and SPN-GLP-1.
Note: Scale bar: 50 nm.
Abbreviations: SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1; GLP-1, glucagon-like peptide-1.

Surface analysis
The key parameters derived from the nitrogen adsorption experiments are listed in . Since both silicon A200 and SPN-GLP-1 were not mesoporous, the surface area here is a more important parameter than pore size. As shown in , the specific surface area of A200 was very large as expected (nearly 200 m2/g) because A200 itself is made of silicon nanoparticles. After loading with GLP-1 and Eudragit® L100, the specific surface area of SPN-GLP-1 decreased to 16.5 m2/g, indicating the adhesion of both the drug and the polymer on the surface of silicon nanoparticles. On the other hand, the data of pore volume and pore size might come from the gaps among particles. Therefore, the smaller particles resulted in smaller pore size, and vice versa.
Table 2 Surface area, pore volume, and pore size analysis by nitrogen adsorption experiment
In vitro release study
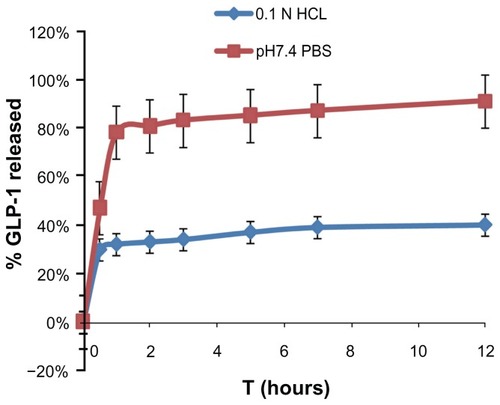
Firstly, a validated HPLC assay was established for the analysis of the peptide drug in vitro and in vivo. Then drug release was studied in two mediums with different pH values. As seen in , the release of GLP-1 from SPN-GLP-1 in pH 7.4 PBS was substantially faster than that in pH 1.0 PBS, which indicated an obvious pH-sensitivity of the nanomatrix system in terms of drug release. It means that less peptide will release in the stomach after oral administration and this is favorable for the absorption of peptide in the small intestine. The cumulative drug released was about 80% and 40% in the two media at the end of test, respectively, but most of the drug released in the first hour. Then the release became slower and the drug release rates were relatively constant after 3 hours. Here, GLP-1 was not used as the control because it is very water soluble and can dissolve in the two mediums immediately.
Figure 4 Release profile of GLP-1 from SPN-GLP-1 at different pH conditions (pH 1.0 and pH 7.4) at 37°C.
Note: Each data point represents the means ± SD of three determinations.
Abbreviations: GLP-1, glucagon-like peptide-1; SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1; SD, standard deviation.
In vivo evaluation
Oral hypoglycemic efficacy of SPN-GLP-1
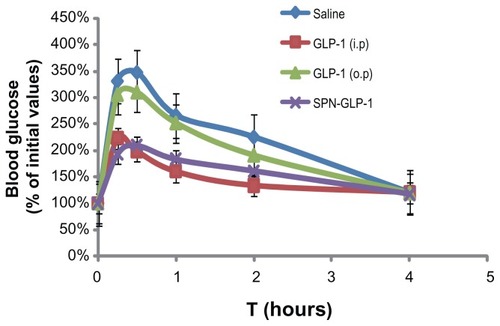
Oral SPN-GLP-1 was compared with oral GLP-1 and ip GLP-1 in hypoglycemic efficacy based on the blood glucose level. As shown in , both the oral SPN-GLP-1 and the ip GLP-1 group in the IPGTT experiment demonstrated a significant hypoglycemic effect compared to the saline group. Oral GLP-1 showed very low efficacy. The hypoglycemic bioavailability (Fp) of the SPN-GLP-1 group was 77% in comparison with the ip GLP-1 group. In other words, the oral administration of SPN-GLP-1 achieved significant hypoglycemic efficacy. In the studies below, oral GLP-1 was not used as a control in vivo because of its low efficacy.
Figure 5 The hypoglycemic efficacy profiles of IPGTT experiment of GLP-1 and SPN-GLP-1 after oral or ip administration at a dose of 1 mg GLP-1/kg to SD rats.
Note: Means ± SD, n = 5 each.
Abbreviations: GLP-1, glucagon-like peptide-1; SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1; IPGTT, intraperitoneal glucose tolerance test; ip, intraperitoneal; SD rats, Sprague Dawley rats; SD, standard deviation.
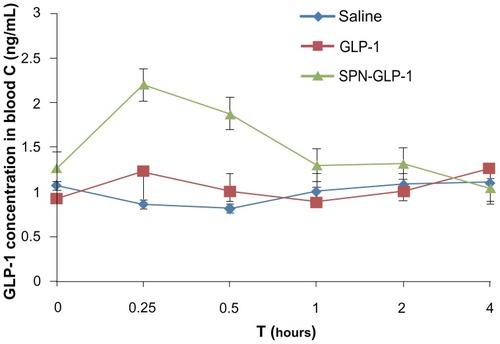
Pharmacokinetics study
The average plasma drug concentration-time profiles of GLP-1 in the rats after a single dose of oral administration of SPN-GLP-1 (1 mg/kg) or ip injection of GLP-1 (0.33 mg/kg) are presented in and . The AUC and Cmax value of GLP-1 in the oral SPN-GLP-1 group were higher than that in the ip GLP-1 group, while the Tmax value for both formulations was similar. The relative bioavailability of the oral SPN-GLP-1 nanomatrix over the ip injection group was 35.67% after taking the dose into consideration, which is quite high for a peptide drug.
Table 3 Pharmacokinetic parameters after oral administration of SPN-GLP-1 (1 mg/kg GLP-1) and ip administration of GLP-1 (0.33 mg/kg) to SD rat
Figure 6 Mean plasma GLP-1 concentration-time profiles following oral administration of SPN-GLP-1 at a dose 1 mg/kg and ip injection of GLP-1 at a dose 0.33 mg/kg to SD rats.
Note: Means ± SD, n = 3.
Abbreviations: GLP-1, glucagon-like peptide-1; SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1; ip, intraperitoneal; SD rats, Sprague Dawley rats; SD, standard deviation.
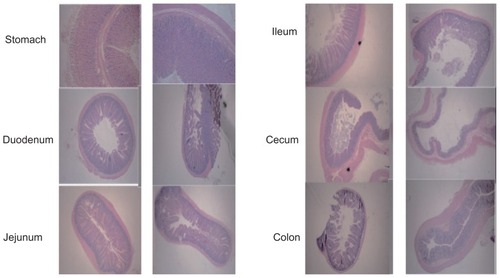
In vivo safety evaluation
The living status, visible toxicity, and any changes in the different gastrointestinal sections of the animals were observed throughout the in vivo studies. After oral administration of SPN-GLP-1, the activities of the animals were similar to those in normal condition. Moreover, hematoxylin-eosin staining results of the stomach, duodenum, jejunum, ileum, cecum, and colon () showed that there were no obvious acute gastrointestinal injuries or morphology changes, which basically proved the safety of the SPN-GLP-1.
Figure 7 Hematoxylin-eosin staining photos of stomach, duodenum, jejunum, ileum, cecum, and colon following oral administration of SPN-GLP-1 at a dose 1 mg GLP-1/kg to SD rats.
Abbreviations: GLP-1, glucagon-like peptide-1; SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1; SD rats, Sprague Dawley rats.
Absorption mechanism
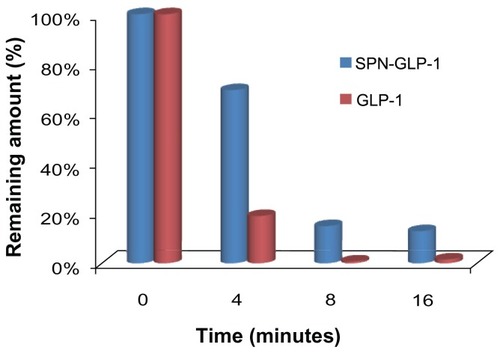
Stabilities of SPN-GLP-1 against intestinal enzymes
The stabilities of the SPN-GLP-1 in intestinal fluid are shown in . It was clear that GLP-1 rapidly degraded with a half-life of 2 ± 0.02 minutes, due to the presence of high concentrations of digestive and brush-border enzymes in the intestinal fluid. In contrast, the SPN-GLP-1 showed significantly greater proteolytic stabilities in the same condition. For instance, about 70% of the drug remained intact in the SPN-GLP-1 group at 4 minutes while less than 20% of native GLP-1 remained. This demonstrated the protective effect of the nanomatrix system on the peptide drug against intestinal enzymes, likely because of the drug adsorption on the silica nanoparticles and the encapsulation of Eudragit® L100 outside.
Figure 8 Stabilities of GLP-1 and SPN-GLP-1 against intestine enzymes.
Notes: Degradation profiles of GLP-1 and SPN-GLP-1 were incubated with rat intestine fluid. Data are presented as means ± SD of three determinations.
Abbreviations: GLP-1, glucagon-like peptide-1; SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1; SD, standard deviation.
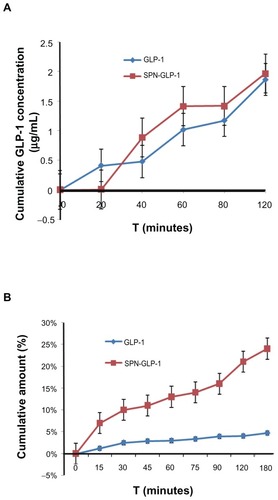
Transport across the mucosa of the intestine
The transports of GLP-1 across Caco-2 cell monolayers and rat gut mucosa are illustrated in . As shown in , there was no significant difference found between GLP-1 and SPN-GLP-1 in the test with Caco-2 cell monolayer; however, the transported amounts of GLP-1 in the SPN-GLP-1 group across the mucosa of the intestine were significantly higher than that of the GLP-1 group (). And the difference between the two GLP-1 formulations increased as time went on. At 180 minutes, 24% and 5% of GLP-1 transported across the intestinal mucosa in the nanomatrix and native drug groups, respectively, suggesting a five-fold mucosa permeability of GLP-1 in SPN-GLP-1 over the control group.
Figure 9 (A) Permeability of SPN-GLP-1 in a Caco-2 cell monolayer transport system. Permeation profiles from the basolateral to apical side. Data are presented as means ± SD of four individual tests. (B) Permeability of SPN-GLP-1 across the mucosa of the intestine by the everted rat gut sac method. Data are presented as means ± SD of three individual tests.
Abbreviations: GLP-1, glucagon-like peptide-1; SPN-GLP-1, silica-based pH-sensitive nanomatrix loaded with glucagon-like peptide-1; SD, standard deviation.
Discussion
Through the combination of silica nanoparticles and a proper pH-sensitive polymer, we designed and prepared a unique system coded as the nanomatrix which is different from the conventional solid dispersion. In solid dispersion, the drug itself was evenly dispersed in the matrix of the polymer in molecules or microcrystals. However, in the nanomatrix system, the nanoscale solid particles were well dispersed in the matrix of the polymer and drug molecules were adsorbed on the surface of these nanoscale solid particles ().
In other words, the difference between these two systems is the use of nanoscale solid particles. As far as we know, the unique features of silica nanoparticles, such as their chemical stability, their safety for pharmaceutical use, and especially their nanoscale structure with large surface areas, have made them ideal for hosting guest molecules of various sizes, shapes, and functions. So the use of nanoscale solid particles may achieve a highly dispersed state for the drug adsorbed on their surface, which is favorable not only for insoluble drugs but also for the soluble macromolecules. On the other hand, the adsorption of the drug on the surface of solid nanoparticles may protect the drug from crystallization which is a tough problem in solid dispersion. Additionally, the addition of such nanoscale solid particles will make the process much easier than the conventional solid dispersion since they themselves are a very good lubricant. Based on a previous test with an orthogonal design experiment (data not shown), A200 was chosen as the nanoscale solid particles. A200 is a silica material with a particle size about 10 nm.
When delivered orally, peptides first encounter pepsin, which is active in the harsh acidic condition (pH 1.2) of the stomach. Therefore, a pH-sensitive polymer material is very useful to reduce the pepsin digestion of loaded peptides. In this respect, Eudragit® L100, a typical pH-sensitive and commercially available polymer was selected in our test based on our previous study.Citation23 Eudragit® L100 is soluble above pH 6.0 and has been used as an enteric coating in pharmaceutical manufacturing for many years. The Eudragit® L100 coating will bypass the stomach environment elevating the fraction of SPN-GLP-1 to reach the intestine. Moreover, formulated together with nanoscale silica particles, Eudragit® L100 will protect the peptide from enzyme degradation to some extent in the GI tract.
We previously found that the nanoparticles made of enteric polymers only obviously increased the oral bioavailability of drugs including insoluble cyclosporine.Citation28 But the preparation process before was complicated and the final product was a colloidal solution which was not stable. With the addition of solid silica particles, it is now very easy to prepare a solid and stable nanoscale drug delivery system.
In the in vitro release experiment as shown in , it is understandable that the drug released rather fast in pH 7.4 PBS, since Eudragit® L100 would dissolve immediately in such conditions and the drug on the surface of the silica nanoparticles would be uncoated. Besides, relatively higher release rates were observed in the initial phase even in 0.1 N HCl medium, which might be caused by the incomplete or uneven coating of the polymer on the surface of the drug loaded silica. As we increased the polymer, the burst release of the drug in an acidic medium decreased, however, further increases of the polymer would result in difficulty of operation. Namely, with the increase of Eudragit® L100, the solid powder finally obtained was very stiff and very hard to be pulverized. On the other hand, it is quite a large challenge to avoid the initial burst release in an aqueous dispersion of a solid particle system. Usually, 30% burst release in a microsphere system is common.Citation29,Citation30 So, a 30% burst release here does not seem very large for our nanostructured system due to the bigger special surface area. Generally, there was a balance between the burst drug release and the operation difficulty. With the present formulation, we could prepare the nanomatrix system more easily and control the burst release at a relatively low level. Additionally, our present delivery system needs to be improved further.
A human intestine epithelial Caco-2 cell monolayer and an everted rat gut sac method were used here to evaluate the biomembrane permeability of SPN-GLP-1. These are well established models for the intestinal brush-border barrier.Citation31,Citation32 Comparing with , it was noticed that SPN-GLP-1 had a slightly higher permeability than GLP-1. However, the transport of SPN-GLP-1 across the mucosa of the intestine was significantly higher than that of GLP-1. We theorize that different enzyme activities in these two models may cause such a difference. Namely in the everted rat gut sac, the enzyme was active enough to degrade more of the native GLP-1.
Our previous study with the pH-sensitive nanoparticles indicated that one of the main reasons for the absorption enhancement of peptide is the enhanced bioadhesion.Citation30 In another report, we showed the protective effect of pH-sensitive Eutragit® on peptide against digestive enzymes.Citation33 Besides, the interaction between a negatively charged cell surface and a positively charged delivery system or drug may also increase the cell uptake.Citation34–Citation36 However, in this study, all external materials were negatively or not charged at pH 7.4 in which the cell uptake was conducted. So these external materials might contribute little to the interaction with a negatively charged cell surface.
An enzyme pool (rat intestine fluid) was used here to mimic an in vivo intestinal enzyme environment. The pool was a digestive enzyme system containing various pancreatic peptidases such as trypsin, chymotrypsin, carboxypeptidase, and elastase.Citation37 The result showed that SPN-GLP-1 had significantly higher proteolytic stability than the native GLP-1. This enhanced enzyme resistance might be one of the critical factors for the better oral GLP-1 absorption. It was also observed that the degradation of GLP-1 formulations in the test condition was rather fast which might be the result of the high concentration of enzymes in the intestinal fluid.
As seen in and , we demonstrated a significant hypoglycemic efficacy and absorption of GLP-1 after oral administration of the nanomatrix system. However, we found that oral native GLP-1 has very low effect on the blood glucose level, so, ip GLP-1 was also used as the positive control in vivo. Hanato et al reported that in IPGTT using rats, a marked improvement of hypoglycemic effects was observed in anionic liposomal formulations of GLP-1 (100 nmol/kg) with a 1.7-fold increase of insulin secretion after intravenous administration as compared to a GLP-1 solution.Citation38 In pharmacokinetic studies, intravenous administration of anionic liposomal formulations of GLP-1 (100 nmol/kg) resulted in a 3.6-fold elevation of serum GLP-1 levels as compared to GLP-1 injection. Additionally, as we noticed in the in vivo study of SPN-GLP-1, it exhibited a higher hypoglycemic efficiency (77%) than its oral bioavailability (35.67%) compared to ip GLP-1. This might be due to the biological property of GLP-1. In fact, the lowering glucose effect of GLP-1 is not dose-dependent. When the concentration of glucose in the body decreases to a certain level, GLP-1 will not function any more as a hypoglycemic agent. In our efficacy test, doses for both formulations were 1 mg/kg, so it is possible that not all of the GLP-1 exhibited its effect after injection, resulting in the above phenomena.
As we know, glucose is given first in IPGTT and then blood samples are taken afterward to determine how quickly glucose clears from the blood. Compared to a simple hypoglycemic test, IPGTT can reflect the level of insulin resistance in diabetes, so it is usually used to test for diabetes with insulin resistance. The hypoglycemic effect of GLP-1 is closely related to insulin resistance, so in our experiments we use IPGTT to evaluate its effectiveness in lowering blood glucose.Citation38
Finally, there were no toxicity effects found in the nanomatrix system as expected since all the materials used here are excipients for pharmaceutical use and no toxic surfactants were added. This is also an important advantage of such a system since many formulations now use surfactants like Cremophor and Tween which increase the concerns of toxicity in long term use as is needed for chronic illness like diabetes.
Conclusion
In summary, a novel nanomatrix system of GLP-1 was designed and prepared here for the first time. It was indicated that such a system possessed a unique nanoscale structure, obvious pH-sensitivity in drug release, and higher proteolytic stability than free peptide. It exhibited high permeability across biomembranes, high bioavailability after oral administration, high hypoglycemic efficacy, and very low toxicity. Through the direct demonstration here in this study and the related discussion, we understood that the nanomatrix system improves the oral absorption and efficacy of GLP-1 by multiple mechanisms. Some of these were shown here, including: (1) a highly dispersed state of the drug due to the adsorption of drug molecules on the huge surface area of solid silica nanoparticles, increasing the interaction opportunity with the absorption site, the intestinal mucosa; and (2) pH-sensitivity of Eudragit® L100 and its coating outside of the drug loaded nanoparticles, which increases the resistance of peptide against the enzyme system in GI tract. Since GLP-1 is expected to trigger a revolution in the treatment of diabetes mellitus, this orally active SPN-GLP-1 might be considered as a potential oral anti-diabetic agent for type 2 diabetes mellitus.
Acknowledgments
This work was supported by grants-in-aid from 973 national key project (2009CB930303) and from NovoNordisk A/S company.
Disclosure
The authors report no conflicts of interest in this work.
References
- WildSRoglicGGreenASicreeRKingHGlobal prevalence of diabetes: estimates for the year 2000 and projections for 2030Diabetes Care20042751047105315111519
- RoglicGUnwinNBennettPHThe burden of mortality attributable to diabetes: realistic estimates for the year 2000Diabetes Care20052892130213516123478
- HolstJJGut hormones as pharmaceuticals. From enteroglucagon to GLP-1 and GLP-2Regul Pept2000931–3455111033052
- SiegelEGGallwitzBScharfGBiological activity of GLP-1-analogues with N-terminal modificationsRegul Pept1999792–39310210100921
- DeaconCFJohnsenAHHolstJJDegradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivoJ Clin Endocrinol Metab19958039529577883856
- EdwardsCMStanleySADavisRExendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteersAm J Physiol Endocrinol Metab20012811E155E16111404233
- JuhlCBHollingdalMSturisJBedtime administration of NN2211, a long-acting GLP-1 derivative, substantially reduces fasting and postprandial glycemia in type 2 diabetesDiabetes200251242442911812750
- HolstJJDeaconCFInhibition of the activity of dipeptidyl-peptidase IV as a treatment for type 2 diabetesDiabetes19984711166316709792533
- ChoiSBaudysMKimSWControl of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic ratsPharm Res200421582783115180341
- GedulinBRSmithPAJodkaCMPharmacokinetics and pharmacodynamics of exenatide following alternate routes of administrationInt J Pharm20083561–223123818291606
- LimSBRubinsteinIOnyükselHFreeze drying of peptide drugs self-associated with long-circulating, biocompatible and biodegradable sterically stabilized phospholipid nanomicellesInt J Pharm20083561–234535018289811
- SinghRLillardJWJrNanoparticle-based targeted drug deliveryExp Mol Pathol200986321522319186176
- KesisoglouFPanmaiSWuYNanosizing – oral formulation development and biopharmaceutical evaluationAdv Drug Deliv Rev200759763164417601629
- JunghannsJUMüllerRHNanocrystal technology, drug delivery and clinical applicationsInt J Nanomedicine20083329530918990939
- MüllerRHKeckCMChallenges and solutions for the delivery of biotech drugs – a review of drug nanocrystal technology and lipid nanoparticlesJ Biotechnol20041131–315117015380654
- GascoMRLipid nanoparticles: perspectives and challengesAdv Drug Deliv Rev200759637737817582649
- MehnertWMäderKSolid lipid nanoparticles: production, characterization and applicationsAdv Drug Deliv Rev2001472–316519611311991
- HosseinkhaniHDNA nanoparticles for gene delivery to cells and tissueInt J Nanomedicine200634416461
- HosseinkhaniHTabataYSelf assembly of DNA nanoparticles with polycations for the delivery of genetic materials into cellsJ Nanosci Nanotechnol2006682320232817037837
- HosseinkhaniHHosseinkhaniMBiodegradable polymer-metal complexes for gene and drug deliveryCurr Drug Saf200941798319149528
- SubramaniaKPathakSHosseinkhaniHRecent trends in diabetes treatment using nanotechnologyDigest Journal of Nanomaterials and Biostructures2012718595
- FriedrichHFussneggerBKolterKBodmeierRDissolution rate improvement of poorly water-soluble drugs obtained by adsorbing solutions of drugs in hydrophilic solvents onto high surface area carriersEur J Pharm Biopharm200;622171177
- JiaZLinPXiangYA novel nanomatrix system consisted of colloidal silica and pH-sensitive polymethylacrylate improves the oral bioavailability of fenofibrateEur J Pharm Biopharm201179112613421658449
- BarrettEPJoynerLGHalendaPPThe determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isothermsJ Am Chem Soc1951731373380
- YounYSJungJYOhSHYooSDLeeKCImproved intestinal delivery of salmon calcitonin by Lys18-amine specific PEGylation: stability, permeability, pharmacokinetic behavior and in vivo hypocalcemic efficacyJ Control Release2006114333434216884808
- BraggioSFerraraASartoriMEvaluation of the role of intestinal and liver metabolism in the conversion of two different ester prodrugs of sanfetrinem to the parent drug in vitro and in vivo using different rat tissues and a surgically prepared rat modelEur J Pharm Sci2002161–2455112113890
- BouërRBartheLPhilibertCTournaireCWoodleyJHouinGThe roles of P-glycoprotein and intracellular metabolism in the intestinal absorption of methadone: in vitro studies using the rat everted intestinal sacFundam Clin Pharmacol199913449450010456292
- ClelandJLBarronLBermanPWDevelopment of a single-shot subunit vaccine for HIV-1. 2. Defining optimal autoboost characteristics to maximize the humoral immune responseJ Pharm Sci19968512134613498961151
- LangerRClelandJLHanesJNew advances in microsphere-based single-dose vaccinesAdv Drug Deliv Rev19972819711910837567
- DaiJNagaiTWangXZhangTMengMZhangQpH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine AInt J Pharm20042801–222924015265562
- ArturssonPBorchardtRTIntestinal drug absorption and metabolism in cell cultures: Caco-2 and beyondPharm Res19971412165516589453050
- NgKYBorchardtRTBiotin transport in a human intestinal epithelial cell line (Caco-2)Life Sci19935314112111278371628
- WangXQDaiJDZhangHZhangXWangJCZhangQAbsorption mechanism of cyclosporine A loaded pH-sensitive nanoparticles in ratsJ Nanosci Nanotechnol2008852422243118572658
- HosseinkhaniMHosseinkhaniHChenYRSubramaniKIn vitro physicochemical evaluation of DNA nanoparticlesInternational Journal of Nanotechnology201188–9736748
- HosseinkhaniHHosseinkhaniMGabrielsonNPPackDWKhademhosseiniAKobayashiHDNA nanoparticles encapsulated in 3D tissue-engineered scaffolds enhance osteogenic differentiation of mesenchymal stem cellsJ Biomed Mater Res A2008851476017688252
- HosseinkhaniHAzzamTTabataYDombAJDextran-spermine polycation: an efficient nonviral vector for in vitro and in vivo gene transfectionGene Ther200411219420314712304
- YounYSChaeSYLeeSKwonMJShinHJLeeKCImproved peroral delivery of glucagon-like peptide-1 by site-specific.biotin modification: design, preparation, and biological evaluationEur J Pharm Biopharm200868366767517904340
- HanatoJKuriyamaKMizumotoTLiposomal formulations of glucagon-like peptide-1: improved bioavailability and anti-diabetic effectInt J Pharm20093821–211111619698772