Abstract
Background:
A circadian rhythm of symptoms has been reported in allergic rhinitis (AR). Severity of all major symptoms of AR, including runny nose, sneezing, and nasal congestion, is typically at its peak in the morning. The objective of this study was to explore the efficacy of the antihistamine and platelet activating factor (PAF) antagonist rupatadine in the morning and evening and to evaluate whether rupatadine provides effective symptom relief throughout the 24-hour dosing interval.
Methods:
A total of 308 patients ≥18 years of age with PAR was randomly assigned to once-daily rupatadine 10 mg, rupatadine 20 mg, or cetirizine 10 mg for 4 weeks in a placebo-controlled, double-blind study. The main outcome was the morning/evening reflective total symptom score (5TSS) over the treatment period. Secondary endpoints included morning/evening reflective nasal total symptom score (4NTSS), individual symptoms, Pdmax1 as percentage of days with daily severest symptom score ≤1, and subject/investigator evaluation of therapeutic response.
Results:
All active groups were significantly more effective than placebo in improving morning and evening evaluations of 5TSS (P < 0.001) and 4NTSS (P < 0.001) at 2 or 4 weeks. At morning evaluation, there was a significant reduction from baseline for 5TSS with rupatadine 10 mg (−36.8%, P < 0.01) and 20 mg (−46.3%, P < 0.01) compared with placebo. Similarly, 4NTSS was reduced significantly more with rupatadine 10 mg (−34%, P < 0.05) and 20 mg (−41%, P < 0.01) compared with placebo. In the cetirizine 10 mg group, the reduction was −32.7% and −32.2% for 5TSS and 4NTSS, respectively, but this reduction was not significant compared with placebo. The percentage reduction was greater at evening than at morning evaluation. 5TSS reduction with rupatadine 10 mg (−40.7%, P < 0.05) and 20 mg (−49.9%, P < 0.01) and cetirizine 10 mg (−40.1%, P < 0.05) was significantly better than with placebo. 4NTSS values for active groups were also significantly improved versus placebo. When individual symptoms were assessed, statistically significant differences for rhinorrhea (P < 0.01), nasal itching (P < 0.01), and sneezing (P < 0.01) were shown in all active groups compared with placebo at morning and evening evaluations. Pdmax1 index was significantly improved for all active groups and the overall efficacy assessed by patients or investigators showed a significant improvement (P < 0.01) versus placebo at 2 and 4 weeks. The incidence of somnolence was significantly greater in all active groups versus placebo.
Conclusion:
The sustained 24-hour action of rupatadine 10 mg provides an effective control of morning and evening symptoms in patients with PAR treated for up to 4 weeks.
Introduction
Many inflammatory diseases exhibit variations in symptoms over time, and symptoms of allergic rhinitis (AR) have been shown to follow a pattern of circadian variation.Citation1 Severity of symptoms of AR is typically greatest in the morning for all major symptoms, including runny nose, sneezing, and nasal congestion.Citation2 Possible etiologies of increased morning symptoms include increased levels of histamine and other inflammatory mediators.Citation3 Patients report that morning symptoms reduce quality of life throughout the rest of the day. Therefore, an important consideration in the pharmacologic treatment of AR is the effective relief of morning symptoms.
Second-generation oral antihistamines are among the most widely prescribed agents due to their effectiveness in the treatment of allergic diseases. Although newer long-acting antihistamine preparations permit once-daily dosing, many patients with AR experience breakthrough symptoms and a reduction of clinical potency at the end of the dosing interval. Most antihistamines demonstrate a peak effect approximately 5 to 7 hours after oral administration, and the duration varies depending on the half-life of parent compound and active metabolites.Citation4
The aim of this study was to examine the efficacy of rupatadine, a new antihistamine H1 and PAF antagonist,Citation5–Citation7 which provides effective symptom relief throughout the 24-hour dosing interval in patients with perennial AR (PAR).
Methods
Study design and treatments assessed
This was a randomized, double-blind, parallel-group, placebo-controlled, comparative study of rupatadine 10 mg (R10), rupatadine 20 mg (R20), and cetirizine 10 mg (C10). All 4 treatments (3 active and placebo) were administered orally in identical tablets each morning within 1 hour after awakening. A total of 61 French medical allergologists and pneumologists participated in the trial. The trial complied with local Ethical Committees and Good Clinical Practice guidelines and local clinical trial regulations. All patients gave their written informed consent before being included in the study.
Inclusion and exclusion criteria
Patients aged ≥18 years old with a diagnosis of PAR for at least 12 months, and with a total nasal symptom score ≥5, were included into the study. During a screening visit, the patients had to show a positive skin prick test (diameter of the papule >3 mm compared with saline solution control, or ≥ than histamine at a 10 mg/mL dilution) at inclusion or within 1 year before inclusion. The allergens used in the prick test are usually related to PAR: house dust mites, cat and dog hair, molds, and feathers. Atopic patients with PAR symptoms and seasonal deterioration during the pollen season were allowed to participate in the study. A normal 12-lead ECG had to be documented at the pre-screening visit with the following requirements: QTc < 430 msec for males, and QTc < 450 msec for females. Women of childbearing age had to show a negative pregnancy test and had to use contraceptive measures during the study.
Patients suffering from nonallergic rhinitis (eg, vasomotor, infectious, or drug-induced rhinitis) or with a negative prick test were not included. Treatments with nasal descongestants in the previous 24 hours, oral antihistamines or disodium chromoglycate (previous week), ketotifen (previous month), topical antihistamines (previous 48 hours), systemic or topical treatment with corticosteroids (except for topical hydrocortisone < 1%), immunosuppressants, or any investigational drug within 2 weeks prior to inclusion, were also considered as exclusion criteria. Other relevant exclusion criteria included abnormal laboratory values of clinical significance; certain conditions that may interfere with response to treatment such as mild asthma treated with inhaled bronchodilators or inhaled corticosteroids >800 μg/day of budesonide or beclomethasone, or with >500 μg/day of fluticasone; obstructive nasal polyps; or hypersensitivity to compounds structurally related to the study drug.
Evaluation of efficacy
Each patient received a diary card for daily recording of symptoms at the start of the treatment. Severity scores for 5 (5TSS) individual AR signs/symptoms: nasal (rhinorrhea, sneezing, nasal itching, and nasal obstruction), and non-nasal symptoms (conjunctival itching) were recorded on the diary card every morning (morning) within 1 hour of awakening and prior to dosing (reflective) and every night (evening) at bed-time approximately 12 hours later. In both the morning and evening symptom severity was assessed over the previous 12 hours (reflective) and scored numerically on a scale of 0–3 with 0 = absent, 1 = mild, 2 = moderate, or 3 = severe. The 5TSS is the sum of the ratings for the individual scores.
The investigators examined the patient’s diary card at each follow-up visit (days 14 ± 3 and 28 ± 3) to check treatment compliance and to provide any advice.
Furthermore, a Pdmax1 index was calculated as the percentage of days during the study for each patient when the score of the daily most severe symptom score was ≤1.
Patient and investigator evaluation of therapeutic response to treatment at 2 and 4 weeks was also assessed. In these 2 follow-up visits, the patient’s and physician’s global evaluation of efficacy was scored numerically on a scale of 0 = worsened, 1 = no change, 2 = slight improvement, and 3 = good improvement.
Evaluation of safety
Treatment safety and tolerability were evaluated according to the incidence and type of adverse events spontaneously reported in the patient’s diary or reported as an answer to the investigator’s question of: “Have you noticed any discomfort during these days” at each visit.
Laboratory safety tests (complete blood count and standard serum chemistry), physical examinations, all performed during the study as well as at the end of the study period, were considered. All adverse events were coded using the WHO Adverse Reactions terminology dictionary, and grouped by treatment.
Statistical analysis
It was calculated that 70 patients had to be included in each treatment group (for a total of 280 patients) in order to show the expected difference between active treatments and placebo of 20% in the main efficacy variable, taking into account a dropout rate of 10% and with a protection level of 0.05 against type I random errors and of 0.2 against type II errors.
Analysis of variance was used to compare treatment groups for the quantitative primary and secondary outcomes. In case of significant results, subsequent pairwise contrasts using a Bonferroni adjustment were made between the treatment groups. For quantitative (efficacy and safety) variables, mean, median, standard deviation, and maximum and minimum values were calculated.
Qualitative variables were expressed as relative frequencies. Chi-square test was used for qualitative variables and Fisher’s test was used if the applicability conditions were not present. The Mantel–Haenszel chi-square test was performed if both variables lay on an ordinal scale.
Analysis of both efficacy and safety was based on intention to treat (ITT), including all patients who were randomized and received at least 1 dose of study medication. The adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA® v 12.1) and the incidence of adverse events was compared between treatment groups using the chi-square test.
All statistical analyses were 2-tailed, with a significance level set at P < 0.05. Statistical analyses were performed using the SAS® statistical software (SAS Institute Inc, Cary, NC).
Results
Study population
Patients were recruited from a total of 61 allergologists and pneumologists from several private centers in France. The disposition of patients during the study is shown in .
Figure 1 Disposition of patients during the study (ITT population).
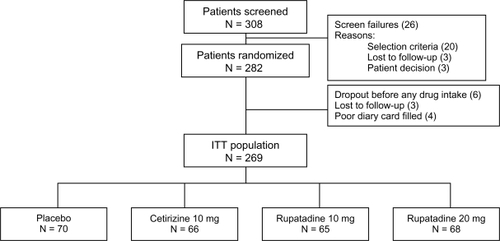
A total of 308 patients were enrolled into the trial, but 26 were not randomized and did not take any study treatment. Therefore, 282 patients were randomized. Of these, 273 took at least 1 treatment dose and were evaluated for safety. Four patients showed incomplete and no valid information in their diaries and were excluded from the data analysis; therefore, the ITT population was 269 patients.
At baseline, demographic data showed there were no differences between relevant demographic and clinical characteristics ().
Table 1 Demographic and clinical characteristics at baseline in ITT population
Efficacy of treatments
and show the ITT analysis for morning and evening score indexes at baseline, and at 2 and 4 weeks of treatment, considering the absolute values and the percentage reduction from baseline.
Table 2 Summary of morning (morning reflective) total symptoms score (5TSS) and nasal total symptoms score (4NTSS) assessments (ITT population)
Table 3 Summary of evening (evening reflective) total symptoms score (5TSS) assessments and nasal total symptoms score (4NTSS) (ITT population)
All active groups showed a significant reduction in symptoms score at morning and evening evaluations, and both 5TSS (P < 0.001) and 4NTSS (P < 0.001) at 2 or 4 weeks showed significant improvements in the ANOVA comparison with placebo.
At the morning evaluations at 4 weeks, there were significant reductions of −36.8% from baseline in the primary endpoint (5TSS, reflective) in the R10 group (P < 0.01) and −46.3% in the R20 group (P < 0.01) compared with placebo. The C10 group reduction of −32.7% from baseline was not significant compared with placebo. At the morning evaluations at 4 weeks, there were also significant reductions in the 4NTSS for both R10 and R20 groups compared with placebo (−34%, P < 0.05 and −41%, P < 0.01, respectively). The C10 group reduction of −32.2% was not significant compared with placebo.
At the evening evaluations at 4 weeks, there was a significant reduction of 5TSS for both R10 and R20 groups (−40.7%, P < 0.05 and −49.9%, P < 0.01, respectively) compared with placebo. The C10 group also showed a significant reduction from baseline (−40.1%, P < 0.05) compared with placebo. The above pattern was similar for 4NTSS scoring: both R10 and R20 groups showed a significant reduction compared with placebo (−40.7%, P < 0.05 and −44.9%, P < 0.01, respectively) and C10 also showed a significant reduction (−39.9%, P < 0.05) compared with placebo.
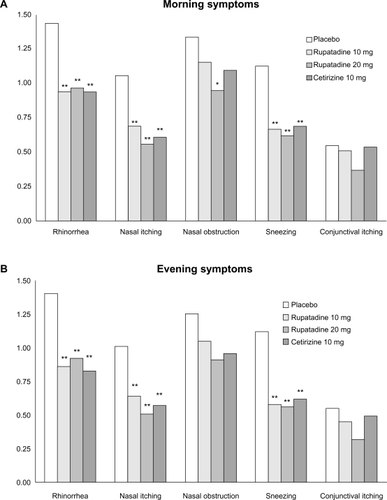
When individual symptoms were assessed, statistically significant improvements in rhinorrhea (P < 0.01), nasal itching (P < 0.01), and sneezing (P < 0.01) were shown in all active groups compared with placebo at morning and evening evaluations. Nasal obstruction was significantly improved only in the R20 group compared with placebo at morning evaluations. A lesser reduction was detected for ocular symptoms at morning and evening evaluations in all active groups compared with placebo ().
Figure 2 Symptom scores: A) Morning evaluation for each individual symptom at 4 weeks; B) Evening evaluation for each individual symptom at 4 weeks.
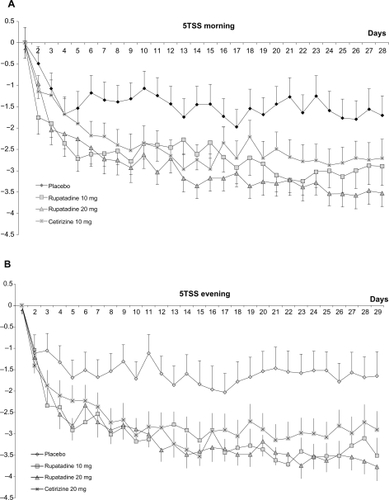
The reduction of 5TSS was also evaluated throughout the study and the circadian rhythm for each treatment group at morning and evening evaluation is shown in .
Figure 3 Evolution of total symptoms score (5TSS): A) morning evaluation for the 5TSS scores during study period; B) evening evaluation for the 5TSS scores during the study period.
All the active treatments were also significantly better than placebo as evidenced by Pdmax1 mean values: placebo = 24.4%; C10 = 43% (P < 0.01); R10 40% (P < 0.01) and R20 49.6% (P < 0.001).
There was a significant difference between treatment groups (P = 0.001) for overall efficacy assessed by patients or investigators at 2 and 4 weeks. All 3 active treatments were evaluated as better than placebo and the differences in pairwise comparisons were statistically significant (P < 0.01) versus placebo at both periods. No difference was found in the comparisons between active treatments.
Safety
presents the incidence of adverse events occurring in ≥1% of patients in any group. There was no significant difference between groups in the number of patients reporting adverse events, or in the total number of reported adverse events. No life-threatening adverse event occurred. The only adverse event showing a statistically significant difference was somnolence: R10 (11%) and R20 (20%) compared with placebo (P < 0.01 and P < 0.001, respectively). Laboratory tests analysis and ECG parameters (QTc interval) did not show any clinical relevant findings between groups.
Table 4 Incidence of adverse events reported by ≥1% during the study by treatment group
Discussion
In the present study, conducted in adults, we have demonstrated that once-daily administration of rupatadine is significantly more effective than placebo in relieving the symptoms of PAR during the 4 weeks of treatment. Moreover, a clear and significant reduction in nasal and non-nasal symptoms score at morning and evening evaluations was seen in rupatadine groups compared with placebo.
The nonsedating H1 antihistamines are important medications in the treatment of all stages of AR severity and are recommended by current guidelines.Citation8 Although newer second-generation antihistamines permit once-daily dosing, many patients experience breakthrough symptoms and a diminution of clinical potency at the end of the dosing interval.Citation4 Over the 4-week follow-up period, the study aim was to evaluate if rupatadine provided a full 24-hour efficacy in our PAR patients.
The symptoms of AR vary in severity over the course of the day and are often worse in the morning. In AR the intensity of nasal congestion, rhinorrhea, and sneezing are greatest early in the morning in approximately 70% of patients.Citation9,Citation10 This was true both for patients with seasonal symptoms alone (55.9%) and for those with PAR (65.7%), although it is noteworthy that those with PAR reported that the worse symptoms were most severe in the morning significantly more often than those with seasonal AR.Citation11 Therefore, to maximize the benefits for patients and to maintain a good overall efficacy and safety profile, any pharmacologic agent used in the management of AR should be effective in controlling these peak morning symptoms. In general, antihistamines would be expected to exert their maximum effect near or shortly after peak serum levels are reached. Previous studies with rupatadine showed a fast onset of action,Citation6 due to the fact that peak serum levels were reached around 0.5 to 1 hour after dosing.Citation7 This was the principal reason that morning dosing was scheduled in our patients. In spite of this fact, we expected to observe the most relief in morning symptoms in comparison with evening symptoms. But overall relief of symptoms was equivalent for morning or evening with rupatadine 10 or 20 mg once daily, indicating that the sustained 24-hour effects of rupatadine are independent of time of dosing. A similar morning/evening profile was observed for cetirizine 10 mg treatment compared with rupatadine 10 mg. Nevertheless, the percentage reduction for both total symptoms and nasal symptoms score was not significant with cetirizine compared with placebo at morning evaluations, whereas the reduction was statistically significant with both rupatadine doses.
When individual symptoms were evaluated at morning or evening, as secondary endpoints, nasal obstruction and conjunctival itching did not show a significant reduction with rupatadine 10 mg and cetirizine 10 mg compared with placebo. Only rupatadine 20 mg provided a significant capacity for alleviating nasal congestion at morning evaluation. Nasal congestion is a particularly troublesome symptom of AR and often is cited by patients as the most bothersome symptom. However, newer antihistamines have demonstrated anti-inflammatory properties, and the results of clinical trials of their effects on nasal congestion are mixed.Citation12 Rupatadine has been shown to reduce effectively nasal congestion in patients with seasonal AR, whether measured objectively as nasal airflow or subjectively as symptoms in allergen exposure study.Citation13,Citation14
The specific mechanisms underlying the chronobiology of AR are speculative; however several factors might contribute to the occurrence of maximum nasal congestion: sneezing rhinorrhea in the morning; secretions increase and accumulate overnight; there is continuous allergen exposure to mold, mites, or house dander; cortisol levels are lowest at night, and hence inflammatory mediators might be at high levels; and autonomic nervous system activity at night promotes vagal tone, favoring vasodilation.Citation15
Differences in daytime and night-time administration of first-generation antihistamines have been reported long time ago and may represent a class effect.Citation16 First-generation H1 antihistamines effectively reduce AR symptoms but worsen daytime somnolence, decrease reaction time, and impair performance.Citation17 Older antihistamines also worsen sleep architecture and disrupt the normal sleep process, and therefore the patients wake up feeling unrested.Citation18,Citation19 Administration time-dependent differences in the pharmacokinetics, especially duration and time to pick effect, between the first-generation antihistamines and the second-generation antihistamines have also been demonstrated.Citation9 The second-generation antihistamines have a rapid onset of action and are not known to interfere with sleep. A study with desloratadine in seasonal AR showed no statistically significant difference in efficacy when the compound was given in the morning or in the evening.Citation2 Rupatadine administered in the morning improves daytime and night-time symptoms, despite rupatadine not being administered at bedtime in this study, and therefore it is impossible to determine chronotherapeutic benefit to evening versus morning dosing of rupatadine.
It is well recognized that second-generation antihistamines are generally nonsedating therapies; however this does not mean that somnolence never occurs with these therapies. Indeed, nonsedating second-generation antihistamines with zero somnolence do not exist. Rather, somnolence is reported in a small minority of patients, which means second-generation antihistamines are nonsedating compared with first-generation ones.
It should be noted that in our trial, at the end of each weekly treatment period, patients were actively asked to report any adverse symptom or event that they may have experienced. This can lead to an ‘over-reporting’ of adverse event frequency in comparison with those studies in which patients have reported adverse events spontaneously. The increase in the incidence of sleepiness, as a treatment-related adverse event, could be associated with the administration of the drug in the morning. The time of drug administration in our study differs from that of other similar trials carried out with other recent second-generation antihistamines, in which the drug in usually taken at bedtime.Citation20
In conclusion, the sustained 24-hour action of rupatadine 10 mg provides an effective control of morning and evening symptoms in patients with PAR treated for up to 4 weeks.
Study participants
Paris: Farid Marmouz (Pontoise), Pascale Beaumont (Saint Maurdes Fosses), Madeleine Epstein (Paris), Dominique Ortolan (Villejuif), Caroline Sauvan (Paris), Dominique Château-Waquet (Paris), Micaela Dona (Paris), Brigitte Medina, (Paris), Véronique Thebault (Aubervilliers), Robert Blassin (Vicennes), Patrice Wurmser (Villeneuve Saint-Georges), Eliane Leriche (Levallois Perret). Bordeaux: Jean-Charles Farauz (Bordeaux), Monique Greciet-Sassoust (Bordeaux), Hervé Masson, (Bordeaux), Philippe Auriol (Bordeaux), Beatrice Michelena (Marmande), Maryse Beau-Besnard (Le Bouscat). Toulouse: Jacques Gayraud (Tarbes), Suzanne Carme (Albi), Jean-Claude Roca (Tarbes), François Malaquin (Albi). Nantes: François Wessel, Anton Michael. Tours: Marie Chantal Carre-Faure, Jean-Philippe Maffre, Charles Truche. Lyon: Françoise Bouteloup, Françoise Roitte-Flandrois (Le Peage de Roussillon), Roch Sanchez (Saint Priest), Marie-Claire Chaize (Venisseux), Lucile Courvoisier (Oullins), Elisabeth Gautier (Lyon), Martine Chouraqui (Lyon). Saint-Etienne: Colette Chappard, Pauline Lazar, Dominique Mounier, Charles Dzviga. Valence: Bernard San Juan. Limoges: Christine Bertin, Marie-Christine Brianchon. Montluçon: Philippe Chalmet. Poitiers: Katy Breuil, Hélène Pouvreau. Angôulême: Jacques Petit, Olivier Lagrange, Ghassan Haddad, Isabelle Bosse (la Rochelle). Nantes: Anne Bataille, Bruno Lebeaupin (Reze). Montpellier: Bernard Lirsac (Perpignan), Nicole Pueach (Lunel), Pierre Coulet, Robert Clavet. Strasbourg: Pierre Braun, Martin Schaller (Colmar), Sami Taieb (Selestat), Claude Schmitz (Colmar). Marseille: Marie-Françoise Fardeau (Les Milles), Jean-Pierre Marie (Aix en Provence), Yann Massabie.
Acknowledgements
We would like to thank Teodoro Sanchez for help with the English editing of the paper.
This study was partially supported by the National Scientific research program of the Spanish Minister of Science and Technology.
Disclosure
Dr F Marmouz declares no conflicts of interests. Dr Iñaki Izquierdo and Josep Giralt are employees of J Uriach y Compañia, S.A.
References
- StormsWWPharmacologic approaches to daytime and nighttime symptoms of allergic rhinitisJ Allergy Clin Immunol2004114SupplS146S15315536446
- HayeRHøyeKBergOMorning versus evening dosing of desloratadine in seasonal allergic rhinitis: a randomized controlled studyClin Mol Allergy200531615644142
- AoyagiMWatanabeHSekineKCircadian variation in nasal reactivity in children with allergic rhinitis: correlation with the activity of eosinophils and basophilic cellsInt Arch Allergy Immunol1999120Suppl 1959910529614
- BruntonSAAllergy management strategies: An updatePatient Care2002SpringSuppl1625
- MerlosMGiralMBalsaDRupatadine, a new potent, orally active dual antagonist of histamine and platelet-activating factor (PAF)J Pharmacol Exp Ther199728011141218996188
- MullolJBousquetJBachertCRupatadine in allergic rhinitis and chronic urticariaAllergy200863Suppl 8752818339040
- KeamSJPloskerGLRupatadine: a review of its use in the management of allergic disordersDrugs20076745747417335300
- BousquetJKhaltaevNCruzAAAllergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen)Allergy200863Suppl 86816018331513
- SmolenskyNHReinbergALabrecqueGTwenty-four hour pattern in symptom intensity of viral and allergic rhinitis: treatment implicationsJ Allergy Clin Inmunol19959510841096
- SchenkelEEffect of desloratadine on the control of morning symtomps in patients with seasonal and perennial allergic rhinitisAllergy Asthma2006 Proc 27:465–472
- BinderEHolopainenEMalmbergHSaloOAnamnestic data in allergic rhinitisAllergy1982373893966890320
- HorakFImpact and modulation of nasal obstructionAllergy200257Suppl 75252812492726
- ValeroASerranoCBartráJReduction of nasal volume after allergen-induced rhinitis in patients treated with rupatadine: a randomized, cross-over, double-blind, placebo-controlled studyInvestig Allergol Clin Immunol200919488493
- StübnerPHorakFZieglmayerREffects of rupatadine vs placebo on allergen-induced symptoms in patients exposed to aeroallergens in the Vienna Challenge ChamberAnn Allergy Asthma Immunol200696374416440531
- MeltzerEODoes rhinitis compromise nigh-time sleep and daytime productivity?Clin Exp Allergy Rev200226772
- ReinbergAGervaisPUgoliniCA multicentric chronotherapeutic study of mequitazine in allergic rhinitisAnnu Rev Chronopharmacology19853441444
- HidmarchIShamsiZAntihistamines: models to assess sedative properties, assessment of sedation, safety and other side-effectsClin Exp Allergy199929133142
- CasaleTBBlaissMsGelfandEGilmoreTHarveyPDHindmarchIFirst do no harm: managing antihistamine impairment in patients with allergic rhinitisJ Allergy Clin Inmunol2003111SupplS835S842
- ChurchMKMaurerMSimonsFERRisk of first-generation H(1)-antihistamines: a GA(2)LEN position paperAllergy20106545946620146728
- BousquetJBachertCCanonicaGWEfficacy of desloratadine in intermittent allergic rhinitis: a GA(2)LEN studyAllergy2009641516152319624554